All published articles of this journal are available on ScienceDirect.

Dyslipidaemia in Rheumatological Autoimmune Diseases
Abstract
Autoimmunity forms the basis of many rheumatological diseases, and may contribute not only to the classical clinical manifestations but also to the complications. Many of the autoimmune rheumatological diseases, including rheumatoid arthritis and systemic lupus erythematosus are associated with an excess cardiovascular morbidity and mortality. Much of this excess cardiovascular risk can be attributed to atherosclerotic disease. Atherosclerosis is a complex pathological process, with dyslipidaemia and inflammation fundamental to all stages of plaque evolution. The heightened inflammatory state seen in conjunction with many rheumatological diseases may accelerate plaque formation, both through direct effects on the arterial wall and indirectly through inflammation-mediated alterations in the lipid profile. Alongside these factors, antibodies produced as part of the autoimmune nature of these conditions may lead to alterations in the lipid profile and promote atherosclerosis. In this review, we discuss the association between several of the rheumatological autoimmune diseases and dyslipidaemia, and the potential cardiovascular impact this may confer.
INTRODUCTION
The complexity and diversity of many rheumatological conditions is often attributed to their underlying autoimmune nature. Autoimmunity contributes to the clinical manifestations, as well as complications of disease and response to treatment. Rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) have been found to associate with an increased risk for cardiovascular disease (CVD) [1-3], resulting in a significantly shortened lifespan. As a consequence, much speculation and research has focused on the role of both traditional and novel, disease specific, risk factors. In the general population, dyslipidaemia has been shown to be one of the strongest predictors of CVD, with elevated levels of low-density lipoproteins (LDL) forming the primary treatment target according to national guidelines [4]. In this review we discuss the association between several of the autoimmune rheumatological conditions (RA, SLE, primary antiphospholipid sysndrome (primary APS), systemic sclerosis (SSc), and primary Sjogrens syndrome (PSS)) and dyslipidaemia, and the potential impact this has on cardiovascular risk, in particular atherosclerotic plaque formation.
ATHEROSCLEROTIC PLAQUE FORMATION: THE ROLE OF LIPIDS AND INFLAMMATION
Coronary artery disease develops due to the formation and rupture of atherosclerotic plaques. The term atherosclerosis covers a spectrum of disease ranging from endothelial dysfunction and fatty streak development, through to the formation and rupture of a mature plaque. The development of atherosclerotic plaques is complex. Inflammation is fundamental to all stages of atherosclerotic plaque [5], with an intense bi-directional interaction occurring between lipids and inflammation. Rheumatological autoimmune diseases are associated with a heightened inflammatory state in varying degrees, thus these processes may be accelerated.
Endothelial dysfunction is the initiating step in plaque development [6]. Healthy endothelium exerts a number of vasoprotective effects such as vasodilation, suppression of smooth muscle cell growth and inhibition of inflammatory responses, thereby helping to protect against atherosclerosis. Nitric oxide mediates many of these effects by inhibiting platelet aggregation and LDL oxidation, as well as opposing the effects of endothelium-derived vasoconstrictors [7]. Endothelial damage occurs when the fine balance between vasoconstrictive and vasodilatory pathways is disrupted. Although endothelial dysfunction is likely to be a multi-factorial process, the major cardiovascular risk factors such as hypercholesterolaemia, hypertension, diabetes and smoking have been implicated via their ability to increase the production of reactive oxygen species [8]. It is postulated that the increase in reactive oxygen species may in turn reduce endothelial nitric oxide (NO) availability [9, 10]. Multiple lipid abnormalities have been associated with endothelial dysfunction. Hypercholesterolaemia has been shown to cause focal activation of the endothelium in medium and large arteries and has been associated with an increased number of monocytes entering the intima [11]. High levels of oxidised LDL (oxLDL) may down regulate endothelial NO synthase (eNOS), thus reducing available NO and restricting coronary vasodilation [12]. High levels of circulating triglycerides (TGs) may also damage the endothelium via their oxidative charge and result in disruption of the normal NO pathway [13, 14]. Studies into the effects of lipoprotein (a) (Lp(a)) have shown elevated levels to be inversely correlated with small artery compliance and NO production [15, 16]. High levels of systemic inflammation may also disrupt endothelial homeostasis via the NO pathway, by reducing the expression of eNOS and increasing the expression of inducible NO synthase producing a net excess of NO [17]. Too much nitric oxide can also be deleterious to the endothelium, thus a very fine balance of NO needs to be maintained in order to preserve and protect the endothelium [18].
Due to the increased permeability of the dysfunctional endothelium, excess LDL infiltrates the artery wall and is retained in the intima by matrix components. LDL then undergoes modification and oxidation, inducing the endothelial cells to express leukocyte adhesion molecules [19] and initiating an inflammatory response in the artery wall [20]. The contribution of oxLDL particles to the development of atherosclerosis is summarised in Fig. (1).
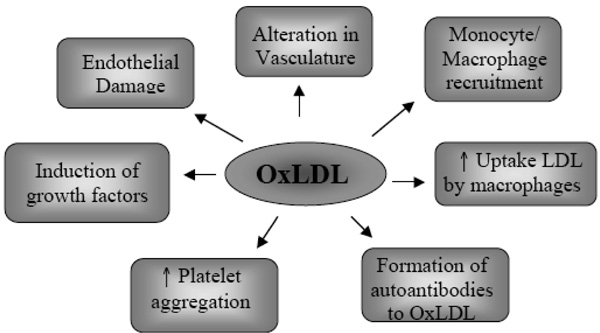
The role of oxidised LDL.

Fatty Streak formation.

Advanced plaque formation.
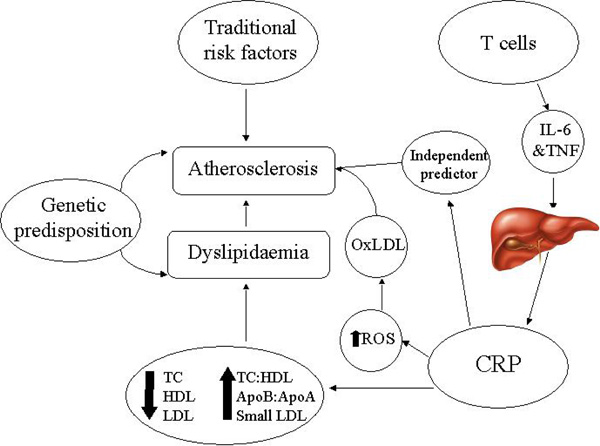
The main mechanisms contributing to dyslipidaemia and atherosclerosis in rheumatoid arthritis. IL-6: Interleukin-6, TNF: Tumour necrosis factor, CRP: C reactive protein, ROS: Reactive oxygen species, OxLDL: oxidised low-density lipoproteins, TC: total cholesterol, HDL: High-density lipoproteins, LDL: low-density lipoproteins, ApoB:ApoA: apolipoproteinB: apolipoproteinA ratio, TC:HDL: total cholesterol: high-desnity lipoprotein ratio.
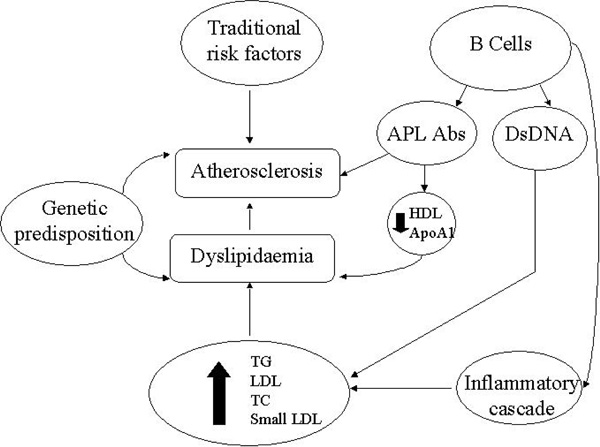
The main mechanisms contributing to dyslipidaemia and atherosclerosis in systemic lupus erythematosus. HDL: Highdensity lipoprotein, ApoA1: apolipoprotein A1, APL Abs: anti-phospholipid antibodies, DsDNA: double stranded DNA, TG:triglycerides, LDL: low-density lipoproteins, TC: total cholesterol.
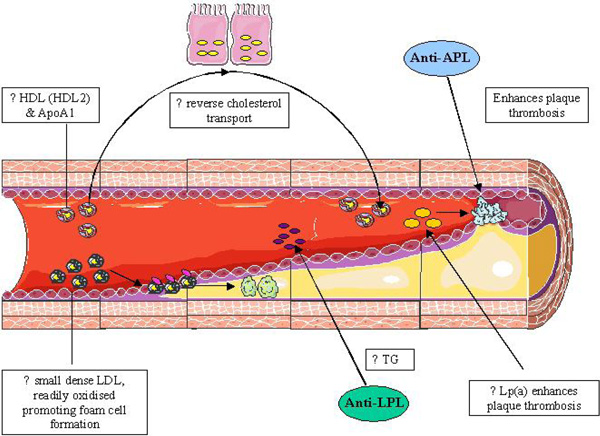
Common changes in the lipid profile amongst the autoimmune rheumatic disease and their impact on atherosclerotic plaque formation. LDL: Low density lipoproteins, TG: Triglycerides, Lp(a): Lipoprotein (a), Anti-LPL: anti-Lipoprotein Lipase, HDL: high density lipoproteins, ApoA1: Apolipoprotein A1, Anti-APL: anti phospholipid.
The first cells attracted to the activated endothelium are platelets [21]. They adhere via glycoproteins on their surface, triggering further endothelial activation resulting in leukocyte infiltration [22]. Once in the sub-endothelial space, monocytes are transformed to macrophages and subsequent incorporation of LDL via endocytosis by scavenger receptors (CD36 and others) differentiates them further into foam cells. The accumulation of foam cells, smooth muscle cells and T cells results in the formation of a fatty streak (Fig. 2a), the earliest recognisable lesion of atherosclerosis [23].
Fatty streaks can progress to form intermediate and advanced lesions (Fig. 2b), by amplification of the processes involved in formation of fatty streaks. The advanced lesions tend to form a protective fibrous cap that walls off the lesion from the lumen. Advanced plaques have a lipid-rich necrotic core containing an abundance of tissue factor, which plays a vital role in thrombus formation upon plaque rupture [24].
Plaque instability and rupture occurs due to uneven thinning or erosion of the fibrous cap. Destabilisation and degradation of the fibrous cap results from the production of inflammatory cytokines, proteases, radicals, coagulation factors and vasoactive molecules, from activated macrophages, T cells and mast cells [25]. The above events lead to plaque rupture, exposing the contents of the core of the plaque to the circulating blood and subsequent activation of the clotting cascade. As the thrombus forms and enlarges, the arterial lumen narrows and ischaemic symptoms may present.
RHEUMATOID ARTHRITIS
RA affects approximately 1% of the adult population in the United Kingdom [26]. RA predominately affects the synovial joints resulting in joint pain, swelling and stiffness, but can also produce an array of clinical manifestations as part of the systemic nature of the disease, ranging from constitutional symptoms to organ involvement. Uncontrolled inflammation can confer a large personal, socio-economic, and psychosocial burden as a consequence of significant joint destruction and deformity.
RA patients have a reduced life expectancy and increased mortality rates compared to the general population [1, 3], with CVD accounting for approximately half of all RA deaths [27]. A recent meta-analysis has shown that the risk of death from CVD is almost 50% higher in RA patients compared to healthy controls [28]. Although CVD in RA can present in many guises (e.g. congestive heart failure, myocardial infarction (MI) and pericarditis), it is those with an underlying ischaemic pathology that pose the greatest mortality risk, as a consequence of atherosclerotic plaque formation [29-31]. Despite this, atherosclerotic disease remains under-diagnosed in RA due to an often atypical or silent presentation [32]. Although the mechanisms responsible for this phenomenon still need to be elucidated, differences in atherosclerotic plaque structure may be responsible. Atherosclerotic plaques of RA patients differ morphologically to those observed in the general population, with less histological evidence of atherosclerosis but far greater plaque instability [33]. Such differences may also offer a potential explanation for the higher fatality rates associated with MI in RA and the increases seen in post MI complication rates [34].
Atherosclerosis in RA may be enhanced through several pathways. Traditional cardiovascular risk factors such as hypertension [35, 36] and obesity [37-39] may occur more frequently in RA or be modified through disease specific mechanisms. In addition, novel risk factors such as systemic inflammation, hyperhomocysteinaemia [40] and activation of the coagulation pathway [41] may further escalate atherosclerotic plaque formation. In fact, the impact of systemic inflammation on CVD is so great it has now been recognised as an independent cardiovascular risk factor in the general population [42, 43]. Although there is a degree of interplay between all of the risk factors, systemic inflammation appears to be fundamental to virtually all. Furthermore, RA patients may be genetically predisposed to the development of atherosclerosis and myocardial infarction [44, 45].
Dyslipidaemia is highly prevalent in RA affecting between 55-65% of patients [46, 47] and can manifest in RA patients with both early [48] and advanced disease [49]. One retrospective study on 1078 blood donors has suggested that the lipid profile may be altered prior to the onset on RA [50]. This study demonstrated that the 79 blood donors who later developed RA had significantly higher total cholesterol (TC), triglyceride (TG), apolipoprotein B (apoB) levels, and lower high density lipoprotein (HDL) levels up to ten years prior to the onset of RA compared to matched controls. This suggests that alterations in the lipid profile may render people more susceptible to the future development of RA [51] or that RA patients are genetically predisposed to the development of RA related dyslipidaemia or that the transcription of these genes is altered by persistent inflammation.
Although dyslipidaemia in RA may be partially governed by a genetic predisposition, it is also influenced by an array of other factors including disease activity [52], reduced physical activity secondary to pain and disability [53], and drug therapy [54, 55]. In untreated RA the lipid profile is characterised by suppression of HDL and TC levels [51, 56, 57]. At first glance this appears to produce a less atherogenic profile, however, HDL levels fall disproportionately more compared to TC levels resulting in an increased atherogenic index (TC:HDL ratio) [48, 56, 58]. The largest study to compare the lipid profiles of untreated RA patients to a control group is the National Health and Nutrition Examination Survey (NHANES III) [54]. This study included 104 patients who were naïve to disease modifying anti-rheumatic drugs (DMARDs) and glucocorticoids (GCs). It confirmed a significant fall in HDL and apolipoprotein A-I (apoAI) levels but failed to demonstrate any significant changes in other lipid levels. Current data exploring the effects of heightened disease activity on TG and low density lipoprotein (LDL) levels is conflicting with some studies reporting a positive association [56] and others a negative association [59, 60]. These discrepancies may be a consequence of limitations of study design, including small study size, differences in baseline characteristics, and incomplete adjustment for confounders.
For many years, apolipoproteins have been recognised as potential markers for CVD in the general population [61]. This is rather unsurprising, as apolipoproteins are found in conjunction with lipid particles (e.g. Apo-AI with HDL and Apo-B100 with LDL). However, emerging evidence now indicates a superiority of measuring apolipoproteins as CVD risk factors, rather than their lipid counterpart (e.g. ApoB rather than LDL) [62]. In RA, Apo-AI levels decrease [48, 52, 54, 63], mirroring the supression in HDL observed in active RA. Although, Apo-B levels have also been reported to be reduced with high levels of inflammation [64] this is to a lesser degree than Apo-AI levels [48, 52, 64].
For many years individual lipid levels have formed the basis of CVD risk stratification algorithms [4, 65]. However, the use of lipid and apolipoprotein ratios is gaining popularity as they have been shown to confer a greater predictive value of first myocardial infarction than individual lipid levels in the general population [66, 67]. The lipid ratios shown to be useful in risk stratification, include TC:HDL ratio [68], LDL:HDL ratio [69] and ApoB: ApoA1 ratio [70]. All three lipid ratios have been reported to increase in active RA [48, 56]. In addition, lipid ratios have been considered as an attractive alternative method of risk stratification in RA as they may overcome the inflammatory mediated fluctuations in individual lipid parameters [71].
Lipoprotein (a) (Lp(a)), is an LDL particle that is bound to apolipoprotein (a), by a disulphide bond with Apo-B100. Current evidence suggests Lp(a) is a key factor in the development of atherosclerosis [72, 73]. The structure of Lp(a) may mechanistically contribute to atherosclerotic plaque formation, through its structural similaririties with plasminogen. Lp(a) competes with plasminogen for its binding site, and stimulates the production of plasminogen activator inhibitor –1 (PAI-1), with the net effect of enhancing plaque thrombosis [74]. In addition, Lp(a) has been shown to interact with other established cardiovascular risk factors [75], including lipid parameters (HDL and LDL) [76]. In RA, several studies have demonstrated a significant increase in Lp(a) levels [48, 54, 56, 77, 78], which in the majority of studies appears to shadow changes in LDL [48, 54, 56]. Although inflammation may partially control these changes in Lp(a), genetic factors may also contribute [79]. Apolipoprotein (a) has several phenotypic variations, due to allelic differences. A recent study has demonstrated that RA patients have a higher frequency of the S3 allele compared to healthy controls (70% vs 39.5%) [79]. The presence of the S3 allele associates with a lower molecular weight of Apo(a) [80]. Furthermore, there is an inverse association between the molecular weight of Apo(a) and Lp(a) levels [81], thus potentially accounting for the increased Lp(a) levels observed in RA.
Further, intricate changes in lipid metabolism have been described in RA. High levels of oxidative stress can occur in conjunction with the chronic inflammatory burden of RA [66]. Lipid particles (in particular LDL) exposed to this environment both within the synovial fluid and the plasma of RA patients are susceptible to oxidative modification [82, 83]. Once oxidised, the LDL particles exhibit enhanced pro-atherogenic properties, through their ability to more readily infiltrate the arterial wall, form foam cells and initiate a localised inflammatory response.
Lipid/lipoprotein particles are not uniform and in healthy individuals are found in a range of sizes and densities. Such diversity is crucial for maintaining the normal pathways of lipid metabolism such as reverse cholesterol transport. However, these structural differences also dictate the atherogenicity of each of the particles. Small dense LDL particles (LDL3) are more prone to oxidation and more readily able to infiltrate the arterial wall than their larger counterparts [84], whereas smaller HDL particles (HDL2) are more successful in performing reverse cholesterol transport and thus confer greater cardio-protection [85]. In RA, the limited available data appears to suggests that RA patients have a more pro-atherogenic lipoprotein subfraction profile with higher levels of small dense LDL particles and lower levels of smaller HDL particles [86]. Alongside this, the balance of pro-atherogenic/anti-atherogenic HDL may be further disrupted through suppression of enzymes including paraoxonase 1 (PON) [87].
In summary, although we have a basic understanding of the effects of RA on the standard lipid profile (TC, LDL, TG and HDL levels) (Fig. 3), our understanding of dyslipidaemia in RA remains far from complete. Further large-scale studies are required to investigate the changes in lipid/lipoprotein structure and function and to establish the relative contribution of dyslipidaemia in its entirety to CVD in RA.
SYSTEMIC LUPUS ERYTHEMATOSUS
Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease that predominately affects premenopausal females, with a peak age of onset between 20-30 years. The condition can manifest with a broad spectrum of clinical signs and symptoms, ranging from relatively minor symptoms such as arthralgia to life-threatening organ involvement. SLE has a relapsing remitting course, often resulting in acute ‘flares’ characterised by a dramatic worsening of clinical symptoms, a heightened inflammatory response and elevated auto-antibody levels.
SLE confers a massive CVD risk, which is far greater than described in other autoimmune diseases. Patients with a disease duration greater than 5 years have been reported to have 52 times greater risk of suffering a myocardial infarction than matched controls [2]. In fact, the processes underlying atherosclerosis are so advanced that subclincal atherosclerosis has even been reported in the paediatric age range [88]. Thus, it is unsurprising that SLE has now been identified as an independent risk factor for the development of endothelial dysfunction, the earliest vascular change in the process of atherosclerotic plaque formation [89]. It is now clear that although traditional risk factors, including dyslipidaemia may be more frequent in SLE these alone do not fully account for the excess cardiovascular morbidity and mortality [90]. Several disease specific factors have been implicated including inflammation, oxidative stress and the presence of anti-phospholipid antibodies [91, 92].
The dyslipidaemia seen in conjunction with SLE is more typical of that described in the general population in relation to CVD, with elevations in TG, LDL, TC and ApoB and a parallel fall in HDL levels [93]. Although the alterations in the lipid profile appear to be exacerbated by active disease, significant changes have been observed in patients with inactive disease [94, 95], thus reflecting that factors other than inflammation may be at play (Fig. 4). Although the lipid profile seen in conjunction with active SLE (elevations in TC and LDL levels) appears to differ from that seen in active RA, there are also similarities (Table 1). Firstly, inflammatory mediators such as CRP and ESR have been shown to suppress HDL levels and increase TG levels [96]. Secondly, other markers of disease activity including the systemic lupus erythematosus disease activity index (SLEDAI) [94], C3d (a specific serological marker of lupus activity) levels [93], anti-double stranded DNA (dsDNA) levels [97] and levels of inflammatory cytokines have been found to associate with lipid levels [98, 99]. One potential inflammation-mediated mechanism that is thought to control some of the lipid alterations seen in SLE, is reduced activity of the enzyme lipoprotein lipase (LPL) [100], which may be governed by circulating inflammatory mediators (e.g. tumour necrosis factor and interleukin-6 (IL-6) [101], and antibodies directed against LPL (anti-LPL) [102]. LPL is responsible for the catabolism of chylomicrons and very low density lipoproteins (VLDL), thus suppression of LPL activity results in an accumulation of TG-rich particles [100]. The presence of cytokines including TNF-α and IL-6 also triggers hepatic production of CRP, which may further exacerbate HDL suppression. The high levels of circulating IL-6 have been shown to inversely correlate with HDL levels.
A Summary of the Classical Lipid/Lipoprotein Profile in Autoimmune Rheumatological Conditions
| TC | HDL | LDL | TG | Lp(a) | oxLDL | TC:HDL | LDL:HDL | ApoB:ApoA1 | |
|---|---|---|---|---|---|---|---|---|---|
| RA | ↓ | ↓↓ | ? | ? | ↑ | ↑ | ↑ | ↑ | ↑ |
| SLE | ↑ | ↓ | ↑ | ↑↑ | ↑ | ↑ | |||
| APS | ←→ | ←→ | ←→ | ↑ | ←→ | ||||
| PSS | ↓ | ↓ | ←→ | ←→ | ←→ | ←→ | |||
| SSc | ←→ | ←→ | ←→ | ↑ | ↑ | ↑ |
RA: rheumatoid arthritis, SLE: systemic lupus eythematosus, APS: anti-phospholipid syndrome, PSS: primary sjogrens syndrome, SSc: systemic sclerosis, TC: total cholesterol, HDL: high density lipoproteins, LDL: low density lipoproteins, TG: triglycerides, Lp(a): lipoprotein (a), oxLDL: oxidised low density lipoproteins, TC:HDL: total cholesterol:high density lipoprotein ratio, LDL:HDL: low density lipoprotein:high density lipoprotein ratio, ApoB: ApoA1: apolipoprotein B: apolipoprotein A1 ratio. ←→ = no change, ↓ = decreased levels, ↑ = increased levels, ? = conflicting data = no data.
High levels of systemic inflammation found in conjunction with SLE result in an increased burden of oxidative stress [103]. Increased oxidative stress can trigger a broad range of pro-atherogenic lipid modifications, including the formation of oxidised LDL (oxLDL), conversion of anti-inflammatory HDL into pro-inflammatory HDL [104], and altering the function of key enzymes involved in lipid metabolism and function [93]. The oxidation of LDL is particularly interesting in SLE, as oxidation affects not only the lipid component of the lipoprotein but also components such as cardiolipin. Antibodies directed against oxidised cardiolipin (anti-phospolipid (aPL) antibodies) are increased in a proportion of SLE patients, predominately those exhibiting features of co-existant anti-phospholipid syndrome (APS) [105]. There are three aPL antibodies found in SLE 1) Lupus anticoagulant 2) anti-cardiolipin antibodies and 3) antibodies to β2 glycoprotein [105]. The presence of aPL antibodies confers an increased thrombotic risk in SLE [106], thus atherosclerotic plaque rupture often has devastating implications due to vessel occlusion as a consequence of massive thrombus formation. The presence of IgG anti-cardiolipin antibodies have also been associated with low levels of HDL and apolipoprotein A-1, thus further escalating CVD risk [107].
SLE patients have high levels of antibodies against oxLDL, which may accelerate the uptake of LDL into the endothelial wall [108]. In addition, there is expanding evidence that antibodies against oxLDL may cross-react with IgM and IgG anticardiolipin antibodies [108, 109], thus further escalating CVD risk. Of particular interest oxLDL forms complexes with β2-glycoprotein I, which have been shown to amplify arterial thrombosis in patients with APS [110], and this may also be a pro-atherogenic mechanism in SLE patients who are positive for APS antibodies.
Several studies have investigated the lipoprotein subfraction profile in SLE, however, the impact of such changes on the development of subclinical atherosclerotic plaque formation and cardiovascular events remains to be established [93, 111, 112]. The first study determined the LDL lipoprotein subfraction profile in 53 patients with SLE and 53 age and sex matched healthy controls, using the method of disc polyacrylamide gel electrophoresis [93]. SLE patients had increased levels of small dense LDL subfractions, which were found to associate with high levels of oxidative stress and elevated CRP levels. Two further studies adopted the method of nuclear magnetic resonance (NMR) spectroscopy, allowing quantification of both HDL and LDL subfractions [111, 112]. The study by Hua et al., examined lipoprotein subfractions in 26 female SLE patients with a history of CVD (myocardial infarction, angina, thromoembolic stroke, or intermittent claudication), 26 age matched SLE patients without CVD and 26 age and sex matched controls [112]. This study demonstrated that SLE patients with a history of CVD had higher levels of small dense LDL than the healthy control population, however the same was not found for SLE patients without CVD. Lower levels of the small ‘cardio-protective’ HDL subfractions were found in SLE patients (with and without CVD) compared to healthy controls. Chung et al. also performed lipoprotein subclassification using NMR spectroscopy in 105 SLE patients (without a history of CVD) and 77 healthy controls [111]. The levels of small dense LDL did not significantly differ between SLE patients and controls and were not found to associate with coronary calcification. The differences observed in the lipoprotein subfraction profile between these studies can be attributed to differences in baseline demographics, the underlying CVD risk, drug therapy and methodology, as well as small study size. Further longitudinal studies are required to elucidate the role (if any) of lipoprotein subfractions to CVD in SLE.
A further two studies have focused on HDL subfractions [113, 114]. Both have demonstrated an atherogenic profile with increased levels of HDL3 and reduced levels of HDL2 compared to healthy controls.
Lp(a) levels have been shown to be substantially increased in SLE patients with studies reporting them to be almost double those seen in matched control populations [115-118]. This appears to be independent of the effects of disease activity, thromobotic events, the presence of aPL antibodies and glucocorticoid use [115]. Furthermore, SLE patients with a history of vascular disease have been found to have higher Lp(a) levels than SLE patients without a history of vascular disease [117, 119], thus indicating a causative role. Unfortunately, all studies to date are limited by the small sample size and cross-sectional design, thus making it impossible to draw conclusions regarding the impact of Lp(a) on CVD in SLE. Similar to the findings in RA, genetic factors are major determinants of elevated Lp(a) levels in SLE, with only the small apolipoprotein (a) phenotypes found to significantly predict Lp(a) levels in SLE [118].
Data regarding changes in apolipoprotein levels and their relative contribution to CVD in SLE is sparse. One study comparing the lipid profile in 53 premenopausal SLE patients to 45 healthy controls reported significant elevations in both ApoA1 and apoB levels, with apoB levels found to associate with the presence of proteinuria [120]. A further study involving 46 SLE patients and 30 matched controls reported conflicting findings, with similar ApoA1 levels observed in both groups [113]. The effects of treatment with glucocorticoids on apoplipoprotein levels also remains a matter of some debate, with one study demonstrating no change in ApoA1 levels [113] and another demonstrating an increase in apoA1 levels [121]. To date there have been no studies assessing the predictive value of apolipoproteins in CVD risk stratification.
ANTI-PHOSPHOLIPID SYNDROME
The Antiphospholipid syndrome (APS) is characterised by a combination of arterial and venous thrombosis, recurrent miscarriages, and thrombocytopenia. Although APS is most commonly found as an overlap syndrome with SLE, it is also recognised as an independent clinical entity (primary APS) [122].
To date, there is much speculation as to whether primary APS patients are at greater risk of atherosclerotic plaque formation than the general population. Some of this uncertainty stems from the significant limitations of the available study data, such as small study size and poor methodology [123], but may also be attributed to the lack of evidence to support a role of APL antibodies in atherothrombosis formation in non-SLE patients with MI [124, 125]. However, several studies on primary APS patients have demonstrated subclinical vascular changes consistent with endothelial dysfunction and early atherosclerotic plaque formation including impaired flow mediated dilatation and increased carotid intima-media thickness [126-128], respectively. Unfortunately, there is a lack of direct epidemiological evidence supporting a higher CVD morbidity or mortality in primary APS.
In line with a relative lack of available information on CVD in primary APS, data on dyslipidaemia in primary APS is also sparse. No studies have specifically been designed to assess dyslipidaemia in primary APS, however, a few small studies have commented on the lipid profile [128, 129]. The first study to comment on the lipid profile in primary APS, did not find any significant differences between TC, HDL, LDL, TG, ApoA1, ApoB, or Lp(a) levels compared to healthy controls [128]. A second study by Medina et al. [129] has reported conflicting findings with a significant increase in TG levels in patients with primary APS compared to healthy controls. These differences can be explained by differences in study size, baseline demographics, inclusion/exclusion criteria and auto-antibody status. Based on these studies alone we are unable to get a true representation of the extent of dyslipidaemia in primary APS (if any) and the impact this may have on CVD, thus further studies are required to address this.
SJOGRENS SYNDROME
Primary Sjogren’s syndrome (PSS) is characterised by chronic inflammation of exocrine glands, resulting in ocular and oral dryness. Although a systemic disease, the inflammation associated with the condition is much less than that seen with RA or SLE. The prevalence of CVD in PSS remains to be established. However, examination of the carotid intima-media thickness of patients with PSS compared to controls has demonstrated a higher prevalence of subclinical atherosclerosis (PSS 49% vs controls 11%) [130]. In this study, the presence of anti-SSA antibodies and leucopenia, but not alterations in the lipid profile, were found to associate with subclincal atherosclerosis.
Two investigators have specifically examined the lipid profile in PSS in relation to inflammation and autoantibody status. The first study conducted by Lodde et al. compared serum lipid profiles in 44 patients with PSS to 12 control patients with oral dryness failing to fulfil the diagnostic criteria for PSS [131]. This demonstrated significantly lower TC and HDL levels in patients with PSS. Other lipid parameters, including lipid ratios (LDL, TG, TC:HDL ratio and LDL:HDL ratio) were not different between patients and control subjects. The levels of TC and HDL in the PSS patients were not found to associate with inflammation (CRP or ESR), however both were found to associate with IgG levels. Alongside this, TC and HDL levels were found to significantly inversely correlate with anti-SSA and anti-SSB antibodies, respectively. A second small study (37 females with PSS and 35 age-matched female controls) confirmed a decrease in HDL levels in patients with PSS, but other lipid components did not significantly differ [132]. HDL levels did not associate with inflammation or auto-antibody status.
Although patients with PSS exhibit some alterations in the lipid profile that are typical of those seen in RA and SLE, including suppression of HDL levels, the mechanisms underlying this need to be examined in more detail. Inflammation appears to be an unlikely culprit due to a lack of association seen in these studies. However, due to the comparative low-grade inflammation seen in PSS much larger scale studies and more sensitive methodology (high sensitivity CRP) may be required to provide the power to detect such a relationship.
SYSTEMIC SCLEROSIS
Systemic sclerosis (SSc) is a condition characterised by skin fibrosis that advances proximally from the distal extremities. In addition, patients with SSc can develop complications arising from organ involvement, including pulmonary fibrosis, renal failure and vasculopathy. The vascular involvement in SSc has traditionally been thought to be due to microvascular disease [133, 134], however, evolving data suggests an increased prevalence of macrovascular disease as well [135]. Patients with SSc have a fourfold increase in mortality compared to the general population, with approximately a third of all deaths due to cardiovascular causes [136]. Although, peripheral vascular disease (PVD) has been reported to be almost 5 times higher amongst patients with SSc than the general population [137, 138], carotid intima-media thickness (IMT), a marker of subclinical atherosclerosis, has been found not to differ significantly between cases and controls [139, 140]. Irrespective of these findings, studies have demonstrated evidence of increased endothelial dysfunction in SSc patients compared to controls, as measured by flow-mediated dilatation [140, 141]. In addition, endothelial dysfunction has been shown to improve following endothelin receptor blockade in SSc [142]. Markers of arterial stiffness such as pulse wave velocity and aortic augmentation index have also been shown to be higher in SSc patients than in a control population [141].
Dyslipidaemia and its contribution to vascular disease/endothelial dysfunction in SSc remains relatively unstudied with only a handful of small studies attempting to address this [135, 139, 143]. The changes in the lipid profile in SSc appear to be subtle, with many of the conventional lipid parameters reported to remain unchanged and only a significant increase in TG levels being witnessed by some investigators [143, 144]. The hypertryglyceridaemia in SSc may be mediated through reduced LPL activity as a consequence of the formation of anti-LPL antibodies [144]. However, when delving deeper it is apparent that further potentially pro-atherogenic changes including enhanced oxidation of LDL [145] and increased Lp(a) levels [146] occur. Increased levels of circulating adhesion molecules have also been noted in SSc. Although these appear to occur in conjunction with endothelial dysfunction [147, 148], to date no direct associations have been made between adhesion molecules and atherosclerotic disease in SSc.
In summary, patients with SSc may have an increased risk of CVD, but this requires further clarification in large-scale studies specifically designed to assess this. In addition SSc patients appear to harbour several alterations in the arterial wall including increased arterial stiffness and endothelial dysfunction, however, the contribution of dyslipidaemia to such changes, if any, requires further investigation.
DRUG THERAPY AND DYSLIPIDAEMIA
The management of many of the autoimmune rheumatological conditions relies upon the use of immunosuppressive agents, which are broadly divided in to glucocorticoids (GCs), disease modifying anti-rheumatic drugs (DMARDs), and biologic agents (e.g. anti-TNF). However, the use of these agents differs significantly between conditions due to efficacy, drug licensing, and side effects. Many of these medications alter the lipid profile with the most widely reported change being an elevation in HDL levels [49, 149-151]. The most likely mechanism to account for such changes in the lipid profile is generic suppression of inflammation. However, some medications have been shown to exert drug specific effects on the lipid profile. For example, Hydroxychloroquine (HCQ), a DMARD used in the management of RA and SLE, has been reported to produce a less atherogenic lipid profile by lowering TC, LDL and TG and increasing HDL levels [152, 153]. The advent of newer ‘potent’ biologic agents offering rapid anti-inflammatory effects has sparked interest surrounding their effect on the lipid profile. However, due to the contraindications of anti-TNF use in SLE patients, most of the data attempting to address this issue has been based in RA patients. The available data demonstrates an increase in TC and HDL levels, with out an overall change in the atherogenic index (TC:HDL) [151, 154]. Thus again potentially reflecting a reversal of the previously inflammatory-mediated suppression of the lipid profile. Despite these findings, very little data exists regarding the more intricate effects of these drugs on lipids, such as lipid subfractions, lipid modifications (e.g. LDL oxidation) and lipid function, and the impact that these may have on cardiovascular risk.
CONCLUSION
Autoimmunity plays a major role in the development of dyslipidaemia and atherosclerotic plaque formation in many rheumatological conditions. The mechanisms underlying these changes include the interplay of inflammation and auto-antibody formation. Although the pattern of dyslipidaemia appears to differ between the autoimmune rheumatological diseases outlined in this review, many of the underlying mechanisms are similar (Fig. 5). Thus treatment options to reduce CVD risk amongst these conditions share a common theme, with the use of disease-modifying anti-inflammatory medications paramount to all.
ABBREVIATIONS
COMPETING INTERESTS
The Authors declare that they have no competing interests
AUTHORS CONTRIBUTIONS
| Tracey E Toms: | Analysis and interpretation of data, drafting manuscript, approval of final version to be published |
| Vasilieos F Panoulas: | Acquisition of data, analysis and interpretation of data, Approval of final version to be published |
| Goerge D Kitas: | Substantial contributions to the conception and design, revision of draft manuscript, approval of final version to be published |
ACKNOWLEDGEMENTS
This work is supported by an Arthritis Research Campaign Clinical Fellowship grant (grant number 18848 to T.E.T), and an Arthritis Research Campaign infrastructure support grant (grant number 17682, given to the Dudley Group of Hospitals NHS Foundation Trust, Department of Rheumatology). Dr Vasileios F. Panoulas is supported by a PhD scholarship from Empirikion Institute, Athens, Greece.

