All published articles of this journal are available on ScienceDirect.

Testosterone and Cardiovascular Disease
Abstract
Cardiovascular disease [CVD] is a leading cause of mortality accounting for a global incidence of over 31%. Atherosclerosis is the primary pathophysiology underpinning most types of CVD. Historically, modifiable and non-modifiable risk factors were suggested to precipitate CVD. Recently, epidemiological studies have identified emerging risk factors including hypotestosteronaemia, which have been associated with CVD. Previously considered in the realms of reproductive biology, testosterone is now believed to play a critical role in the cardiovascular system in health and disease. The actions of testosterone as they relate to the cardiac vasculature and its implication in cardiovascular pathology is reviewed.
1. INTRODUCTION
Cardiovascular disease [CVD] is the leading cause of mortality globally. It was responsible for an estimated 17.5 million deaths in 2012, which accounted for 31% of the global crude death rate [1].
CVDs primarily affect the cardiac parenchyma or vasculature [2]. They are precipitated by non-modifiable as well as modifiable risk factors. Emerging risk factors, such as hypotestosteronaemia have recently been suggested to contribute to the pathogenesis [3, 4].
The majority of CVD aetiologies can be attributed to atherosclerosis. Atherosclerosis, a specific type of arteriosclerosis, which is characterised by lipid accumulation and mononuclear leukocyte infiltration in the tunica intima of vessels, results in mural thickening [5]. Although the exact mechanisms are not fully understood, in most cases oxidised LDL [oxLDL] is often considered the culprit since it is believed to trigger a chronic inflammatory response [6, 7]. The corollary is monocyte and T-cell recruitment to the ‘site of injury’, where monocytes are subsequently transformed into lipid-laden macrophages foam cells along with smooth muscle cells [SMCs] and fibroblasts [8].
Eventually, atheromatous plaque formation occurs and later devlops a necrotic core and fibrous cap [8]. Luminal stenosis consequently limits perfusion. Complications of atheroma include plaque rupture and resultant thromboembolism, plaque fissure, and plaque ulceration [9, 10]. Acute coronary syndromes [ACS] namely ischaemic stroke, unstable angina and myocardial infarction are also sequlae [9, 11, 12].
In 2009 the total cost of CVD per capita from all European Union member states was €212, 9% of the total healthcare expenditure [13]. CVD is therefore a pressing public health issue.
2. TESTOSTERONE AND THE CARDIOVASCULAR SYSTEM
2.1. Testosterone Physiology
Testosterone is an androgen steroid hormone derived from cholesterol [14]. It circulates in two major forms: dehydroepiandrosterone sulphate [DHEA-S] and androstenedione [AS] [15]. Synthesis of testosterone and 5a- dihydrotestosterone [5a-DHT] occurs primarily in the Leydig cells in the testis [15]. Peripheral sites of synthesis include the zona reticularis of the adrenal cortex [DHEA-S and AS] in both sexes, and skin [testosterone and 5α-DHT] to a lesser extent [15, 16]. Testosterone production also occurs in females from Graafian follicle theca interna cells in the form of AS, the majority of which is converted to 17β-estradiol by the enzyme aromatase [17, 18]. Peripheral testosterone production is regulated by hypothalamic gonadotrophin-releasing hormone [GnRH] and the anterior pituitary hormone adrenocorticotropin horomone [ACTH] [16, 19]. On the other hand, gonadal synthesis is governed by GnRH and luteinising hormone [LH] [19, 20]. In both instances, feedback loops exist to ensure adequate regulation of the hypothalamic-pituitary-gonadal [HPG] axis as well as the hypothalamic-pituitary-adrenal [HPA] axis [21, 22].
2.2. Classical Functions
Testosterone acts via the androgen receptor [AR] to mediate an extensive array of physiological processes [23]. Primarily, testosterone is a sex hormone that determines and regulates the development of male secondary sexual characteristics [24]. It increases libido and improves erectile function, but also has a number of anabolic roles. Testosterone stimulates nitrogen retention by increasing the rate of protein synthesis and decreasing the rate of protein catabolism. It is therefore responsible for increasing muscle mass [25]. In addition, testosterone indirectly inhibits bone resorption through its conversion to estradiol [26]. DHT directly stimulates osteoblastic activity and bone growth [27]. In addition to these classical functions, testosterone is also responsible for mediating other crucial physiological processes.
2.3. Non Classical Functions
Testosterone is seldom affiliated with the circulatory system, if at all. Its role as a promoter of erythropoiesis has not extensively been discussed [28]. In fact, an elevated haematocrit is a side effect associated with administration of acute supraphysiological concentrations of testosterone [29]. Furthermore, testosterone has been implicated in cerebral physiology, though this is not without controversy. Testosterone has been shown to improve cerebral perfusion and thus mood in hypogonadal men [30, 31]. Some investigators even go as far as stating that testosterone plays significant roles in memory, strategic planning, higher order motor actions, as well as cognitive and emotional behaviours [24].
2.4. Testosterone and Cardiac Vasculature
2.4.1. Epidemiology
Hypogonadism, as defined by The Endocrine Society’s Clinical Practice Guideline for Testosterone Therapy in Adult Men with Androgen Deficiency Syndromes, is “a clinical syndrome that results from failure of the testis to produce physiological levels of testosterone [androgen deficiency] and a normal number of spermatozoa caused by a disruption of one or more levels of the hypothalamic-pituitary-gonadal [HPG] axis” [32]. The normal testosterone laboratory reference range is 10.4 – 34.7 nmol/L [300 – 1000 ng/dL] [33]. Epidemiological studies have associated low testosterone levels with all-cause mortality and an increased risk of CVD, both of which were statistically significant [4, 34-36]. The association with increased risk of CVD persists even when corrected for age, as testosterone levels are known to increase at approximately age 30, and thereafter decrease with an increase in age [37-39]. In fact, a case control study conducted over a 7 year period, consisting of 2314 men aged 40-79 years, found that every 173 ng/dL [6 nmol/L] increase in serum testosterone was associated with a 21% lower risk of all-cause mortality after correcting for age, body mass index, systolic blood pressure, cholesterol, cigarette smoking, diabetes, alcohol intake, physical activity, social class, education, and sex hormone-binding globulin [35].
While some investigators have linked hypogonadism to an increased risk of coronary artery disease [CAD] others have failed to replicate this association [4, 40]. This discrepancy could be due to differences in study design. Some investigators failed to account for the subfraction of testosterone utilised and the serum quantification method [4]. Testosterone circulates in blood as bioavailable and bound testosterone, and both these subfractions constitute total testosterone [4]. Bioavailable testosterone makes up approximately 33% of circulating testosterone, 30% of which circulates bound to albumin and 1-3% free in blood [4]. On the other hand, the bound testosterone fraction circulates in blood bound to Sex Hormone Binding Globulin [SHBG] and is responsible for 68% of circulating testosterone [4].
Testosterone Replacement Therapy [TRT] serves to substantiate the importance of testosterone in the cardiac vasculature in health and disease (Fig. 1). Three randomised placebo-controlled trials have shown myocardial ischaemia improvement in men with CAD, due to an extension of time to exercise-induced ST-segment depression, as measured on treadmill exercise stress test [41-43]. Moreover, to the best of our knowledge, there is currently no published study in the literature demonstrating a statistically significant association between TRT and cardiovascular adverse events in a large cohort.
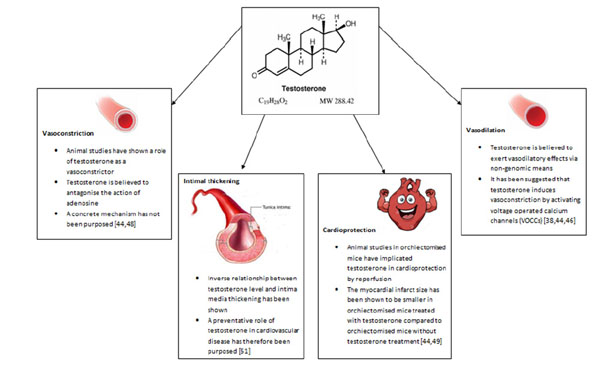
Summary diagram of the key roles of testosterone in cardiovascular system.
Most of the negative connotations attributed to the effects of androgens on the cardiovascular system stem from the observational link established between sudden death and androgen abuse in athletes [24, 38]. To the best of our knowledge, there is no study in the scientific literature presenting statistically significant data to support a causative role for anabolic androgens in cardiovascular adverse events in athletes. In this context, perhaps the chronic administration of anabolic androgens at supraphysiological rather than physiological levels leads to toxicity and death, though robust studies are needed to substantiate this.
2.4.2. Vasodilation
Testosterone is essential for normal healthy vascular function [4]. The consensus from animal studies points to its role in vasodilation [38, 44]. Deenadayalu et al. conducted a study in pre-contracted swine left anterior descending coronary arteries [LAD] where prostaglandin served as the contractile agent. The effects of endothelial denudation and either Krebs-Henseleit Bicarbonate [KHB] solution, or N-nitro-L-arginine methyl ester [L-NAME] on testosterone-treated vessels were tested. KHB is a well-known physiological buffer whereas L-NAME antagonises the enzyme responsible for nitric oxide production, nitric oxide synthase [NOS]. An increase in vascular relaxation percentage and vasodilatation was observed following testosterone administration in a dose-dependent fashion. This effect was not blunted by vascular denudation nor administration of L-NAME and KHB [44, 45]
There is ample evidence in the scientific literature on the vasodilatory effects of testosterone on the vascular system, though little is known about the mechanisms through which testosterone exerts this effect. It has been suggested that testosterone may trigger vasodilatation via endothelial-independent non-genomic means, given its rapid onset, though potentiation by genomic means via the classical AR cannot be excluded [38, 44, 46]. Inhibition of voltage-operated calcium channels [VOCCs] and/or activation of potassium channels [KCs] on smooth muscle cells [SMCs] is believed to be instrumental. Testosterone binds the same domain as nifedipine on VOCCs and thus promotes vasodilation by inactivating VOCCs. An alternate postulate is the activation of voltage-operated potassium channels by testosterone and/or activation of calcium-activated potassium channels which give rise to hyperpolarisation, due to potassium efflux. It has also been suggested that testosterone can inhibit calcium influx through store-operated calcium channels [SOCCs] by blunting the response to prostaglandin F2α [PGF2α]. In terms of testosterone-mediated genomic-dependent mechanisms of vasodilation, it has been hypothesised that testosterone upregulates the expression of endothelial nitric oxide synthase [eNOS], the enzyme responsible for synthesising nitric oxide [NO]. NO in turn brings about relaxation of SMCs and subsequent vasodilatation by activating guanylate cyclase which produces cGMP, which in turn activates cGMP-dependent protein kinase [PKG]. PKG phosphorylates and activates sarcoplasmic reticulum-ATPase [SERCA] which results in augmentation of calcium levels in the sarcoplasmic reticulum [38].
2.4.3. Vasoconstriction
Aside from its suggested role in vasodilation, there are a few basic studies that seem to implicate testosterone in vasoconstriction. A study carried out by Ammar et al. utilised male rabbits that were administered either 25mg/kg intramuscular testosterone weekly or no treatment, followed by the removal of the descending thoracic aorta. The vessel was then either contracted with 5-hydroxytryptamine [5-HT] or dilated with sodium nitroprusside [SNP]. This study showed that aortas from the testosterone-treated animals displayed a higher contractile percentage when exposed to 5-HT and a lower relaxation percentage when exposed to SNP, suggesting that testosterone limits vasodilatation and increases vasoconstriction. The aforementioned observations were statistically significant [44, 47].
A preliminary study conducted by Ammar et al. determined the effects of testosterone on isolated as well as perfused coronary arteries. The vasoconstricitve actions of testosterone were apparent [48]. It is believed that testosterone antagonises the vasodilatory effects of the vasodilator adenosine through increased vascular resistance and competes with other vasodilators, though a concrete mechanism has not been proposed [44, 48].
2.4.4. Cardioprotection
One study investigated the role of testosterone in organ reperfusion following vascular occlusion. Tsang et al. isolated rat hearts from control, orchiectomised, and orchiectomised plus testosterone treatment [2mg/kg daily for 8 weeks] groups. The coronary arteries were pre-occluded for 30 minutes and subsequently reperfused with a physiological solution for 120 minutes. Following isolation, it was observed that when compared to the control group, MI infarct size was largest in the orchiectomised group, compared to the orchiectomised plus testosterone treatment group. This finding suggests that testosterone may have a role in vascular reperfusion through which it can impart cardioprotection [44, 49].
2.4.5. Intima-Media Thickness
Intima media thickness [IMT] is a feature of atherosclerosis [50]. Demirbag et al. conducted a study in 42 male patients without a history of atherosclerosis, who were referred for transoesophageal echocardiography [TOE]. IMT was evaluated by TOE and testosterone levels quantified. The authors discovered an inverse relationship between total testosterone and intima media thickness in the abdominal aorta independent of cardiovascular disease. The results of the study therefore imply a preventative role of endogenous testosterone in coronary artery disease [51].
2.4.6. Gender Differences
The gender differences in testosterone physiology and plasma testosterone concentration in adolescent males and females cannot be ignored. According to Mayo Medical Laboratories, the SHBG-bound testosterone reference range in adolescent males and females at the pinnacle of pubertal development [Tanner stage V] suggests that there is a higher percentage of SHBG-bound testosterone in females [18-144nmol/L or 518-4149ng/dL] compared to males [10-57nmol/L or 288-1642ng/dL]. The physiological implication of this difference is yet to be unravelled. We propose a reservoir-like role for SHBG-bound testosterone to replenish circulating oestradiol in females via the actions of aromatase.
Some investigators believe that testosterone may be more harmful in females than in males [52-55]. This was echoed by an animal study that utilised rat aortic rings from either atherosclerotic tunica intima or healthy tunica media, as well as cultured rabbit SMCs. Female rat aortic rings treated with testosterone resulted in significant decreases in prostacyclin [PGI2] synthesis whilst PGI2 synthesis in male rat aortic rings remained unchanged. It was suggested that testosterone may induce atherogenic changes in females by modulating PGI2 synthesis [56].
The beneficial effects of testosterone in males were also highlighted in another study where a group of testicular feminised mice [Tfm] and littermates were fed a cholesterol-rich diet for 28 weeks. They were then treated with exogenous testosterone alone, exogenous testosterone plus the oestrogen receptor α-antagonist fulvestrant, and exogenous testosterone plus the aromatase inhibitor anastrazole. It was found that exogenous testosterone replacement inhibited fatty streak formation in Tfm mice independent of the androgen receptor. Moreover, it was suggested that the observed HDL-C increment may have been attributed to the conversion of testosterone to 17β-estradiol [57].
It is also important to consider how testosterone can potentiate the effects of CVD risk factors such as obesity in males. It was found that obese men had lower levels of total and free testosterone, as well as SHGB-bound testosterone, possibly due to upregulated aromatase activity which converts testosterone to oestradiol [58]. These individuals were concluded to be at a greater risk of developing CVD and impotency when compared to the control population [58]. Since the SHBG-bound testosterone levels were low, one may argue that increasing levels of SHBG-bound testosterone and concomitantly inhibiting aromatase may increase the circulating levels of free testosterone due to ‘testosterone unloading’, which may help to reduce the risk of impotency and CVD. Therefore, comparing testosterone physiology in males and females may help elucidate further mechanisms by which testosterone exerts its effects, as well emphasizing the sex-specific pathophysiological mechanisms that implicate testosterone in CVD.
CONCLUSION
The scientific consensus from animal studies is vasodilatation, as studies supporting a role for vasoconstriction are limited and conflicting. The reported differences may be attributed to study design. The testosterone concentration utilised differed from study to study, as well as vascular exposure time to testosterone. It is logical to propose that depending on whether exposure to testosterone is acute or chronic, vasoconstriction or vasodilatation may be elicited to maintain vasomotor tone and haemodynamic homeostasis. It is also worth stating that the animal studies were carried out in different species and therefore species-dependent responses may have contributed to the observed differences. More importantly, the limitation of basic science studies is that the animal physiology is not always representative of human physiology, and may thus pose problems when research is translated from ‘bench to bedside’.
The beneficial effects of testosterone in certain scenarios have meant that efforts have been made to explore possible avenues of increasing circulating testosterone concentrations. It was shown that endogenous insulin can help to increase testosterone synthesis and inhibit testosterone binding to SHBG, thereby decreasing concentrations of SHBG-bound testosterone and increasing free circulating testosterone [59]. Nevertheless, one has to consider the long-term effects of chronic insulin administration in non-diabetics as well as the issue of compliance, since insulin can only be administered intravenously due to risk of digestion. Furthermore, the study is limited due to a small study population size.
There are unanswered questions on whether or not men over the age of 50 with coronary artery disease should also be screened for hypogonadism. The difficulty lies in the fact that the role of hypotestosteronaemia in CVD whether causal or consequential, has not been established. If indeed a causal relationship exists then chronic low dose TRT may prove beneficial in clinical trials as this may improve cardiovascular outcome. It is also important to mention that no uniform definition exists for hypogonadism and thus different investigators define it differently in terms of the reference range. Introducing some standardization and consistency to androgen reference ranges that define hypogonadism and hypotestosteronaemia will help to shed light in terms of study design and targeting the right population for investigation and treatment [60-62].
All in all, there is emerging evidence to support the hypothesis that low testosterone levels predict a poor cardiovascular risk profile. Historically considered in the realms of reproductive biology and puberty, it is now clear that the roles of testosterone extend beyond that anticipated. Although we are still far from unmasking the roles and mechanisms of testosterone in the cardiac vasculature, one thing remains certain; that the future is bright. Testosterone holds great promise; possibly the hormone that will revolutionise the field of cardiovascular physiology.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
ACKNOWLEDGEMENTS
Declared none.

