All published articles of this journal are available on ScienceDirect.

Type F Congenital Quadricuspid Aortic Valve: A Very Rare Case Diagnosed by 3-dimenional Transoesophageal Echocardiography
Abstract
Congenital quadricuspid aortic valve (QAV) is a rare cardiac anomaly. Several different anatomical variations of a quadricuspid aortic valve have been described. Aortic regurgitation is the predominant valvular dysfunction associated with QAV and patients tend to present in their 5th or 6th decade of life. This anomaly is rarely picked up by transthoracic echocardiogram (TTE). A comprehensive transoesophageal echocardiography (TOE) study is more likely to diagnose it. We describe a very rare type of QAV – Type F in a 52-year-old lady who presented with symptoms of shortness of breath and pre-syncope. We include TOE images and intra-operative valve images.
INTRODUCTION
Quadricuspid aortic valve is a rare cardiac malformation, with an estimated incidence of 0.0031 to 0.043% of all congenital heart disease. It usually appears as an isolated congenital anomaly [1-3]. The Mayo Clinic has noted an incidence of 1% in a review of patients undergoing surgery for pure aortic regurgitation [4]. However, QAV may also be associated with other malformations, the most common being coronary artery anomalies [5,6]. QAV was mainly an incidental finding at open-heart surgery or at autopsy in the past [7].
CASE REPORT
A 52-year-old lady presented to our centre for echocardiogram from her primary-care-physician because he elicited a cardiac murmur. She had history of worsening breathlessness over a period of 7 years. She also had a history of pre-syncope.
She had a standard transthoracic echocardiogram (TTE), which revealed that her left ventricle (LV) is dilated with evidence of significant systolic dilatation (LV internal diameter systolic 55 mm) associated with severe systolic impairment with an ejection fraction of 33% (Simpson’s Bi-plane method). The aortic valve appeared to be bicuspid on sub-costal views. The parasternal views of the aortic valve were somewhat limited. There was evidence of severe aortic regurgitation (AR) with a central regurgitant jet, which occupied greater than 65% of the LV outflow tract and there was clear flow reversal in the aortic arch and descending aorta. In addition to these findings there is also mild mitral regurgitation (MR) and preserved right ventricular (RV) systolic function. She also informed us that mother had aortic root pathology and had to have aortic root replacement surgery. This hinted to hereditary congenital aortic or aortic valve pathology. She was admitted to our unit and received heart failure treatment and consequently had transoesopha-geal echocardiography (TOE) (Philips IE33 using 3D-TOE probe).
TOE ACQUISITION AND RESULTS
In aortic short-axis views she has quadricuspid aortic valve with 4 sinuses. We identified that she had 2 equal larger cusps and 2 unequal smaller cusps, consistent with type F QAV (Figs. 1, 2). 3D zoom data was acquired in the AV long-axis views. TOE confirmed the finding of severe AR (Fig. 3). This 3D data was later edited on offline QLAB Software tool to acquire aortic valve views from the aortic root (Fig. 4, Video 1).

2-Dimensional diastolic short-axis view of quadricuspid aortic valve on TOE.
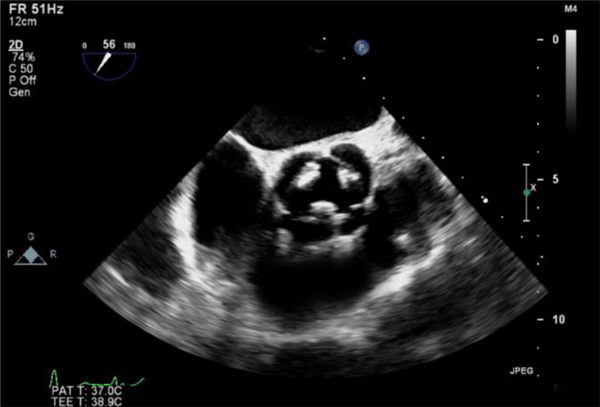
2-Dimensional systolic short-axis view of quadricuspid aortic valve on TOE.
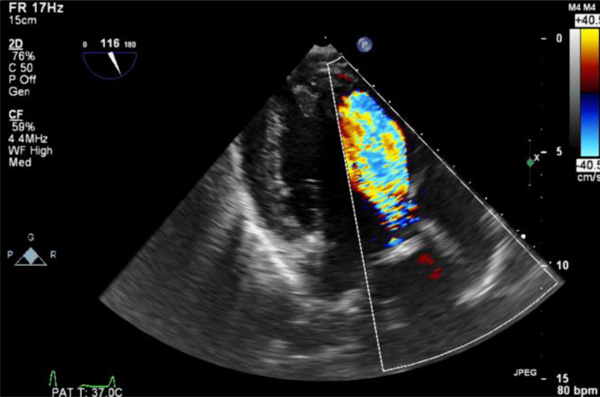
Trans-gastric colour doppler view of severe aortic regurgitation.
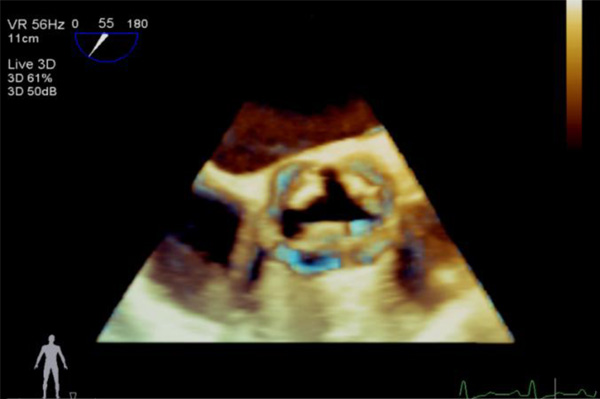
3-Dimensional view of aortic valve from the aortic root clarifying the anatomy of quadricuspid aortic valve.
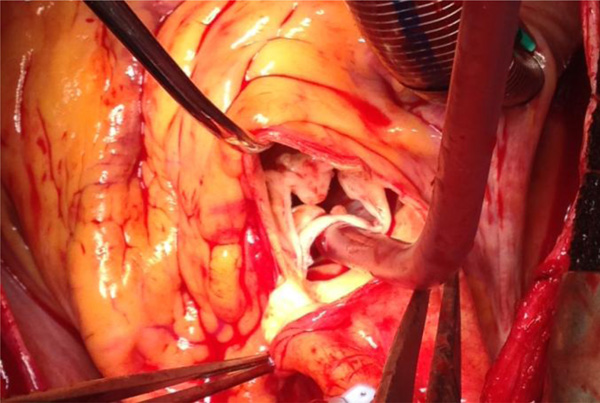
Intra-operative view of the quadricuspid aortic valve.
Once she was stabilised with heart failure treatment, she had prompt surgery to replace her aortic valve (Fig. 5). She made un-eventful recovery and was discharged one-week after AV-surgery.
LITERATURE SEARCH
We carried out a literature review to find how many type F cases have been reported on MEDLINE. Searches were carried out using the following words – ‘quadricuspid AND aortic AND valve AND type AND F’. Three case reports were identified [8-10]. Two cases were mainly diagnosed as part of work-up for surgical repair for aortic incompetency [8, 9]. One case was of a 26-year old woman who presented with infective endocarditis and was diagnosed with type F QAV on TOE [10]. We did not identify any case, which had heredity link with aortopathy or any aortic valve disease.
DISCUSSION AND REVIEW
Recent advances in non-invasive cardiovascular imaging have enabled us to define the anatomy of QAV with high accuracy. Around 200, mainly adult cases have been described in the literature. A major review included 184 patients, with a slight male predominance [5]. Case reports or series of children being diagnosed with this congenital heart anomaly are generally small [11]. More than half of the patients with this abnormality will develop aortic insufficiency in adulthood. It is vital that patients with QAV who present with progressive AR undergo valve replacement or repair at appropriate time. Hence, it is important to promptly detect this malformation and arrange appropriate investigative follow up.
CLASSIFICATION OF QUADRICUSPID AORTIC VALVE
According to the anatomy of the 4 cusps, Hurwitz and Roberts categorized QAV into 7 subtypes (A to G) (Table 1). [1]. Almost 85% of cases are of type A, B or C [1].
Hurwitz and Roberts’ anatomical classification of QAV. (Permission for re-prints obtained from Elsevier Limited, license number: 3293700398552).
| Type | Description |
|---|---|
| A | Four equal cusps |
| B | Three equal cusps and one smaller cusp |
| C | Two equal larger cusps and two equal smaller cusps |
| D | One large, two intermediate and one small cusp |
| E | Three equal cusps and one larger cusp |
| F | Two equal larger cusps and two unequal smaller cusps |
| G | Four unequal cusps |
The morphogenesis of QAV is unknown in man. However, an animal case-report supported the hypothesis that QAV result from the division of 1 of the 3 mesenchymal anlagen that give rise to the normal aortic valves [12].
The physiopathology of the QAV dysfunction is poorly understood. Anatomical abnormalities of the cusps could induce unequal shear stress leading to fibrotic changes and incomplete coaptation [2]. However, insufficiency is also observed in cases of quadricuspid valve with 4 equal cusps [2]. As in bicuspid valve, regurgitation of an abnormal aortic valve may also occur as a result of a prolapse of the layer of the cusps or be associated with aortic root dilatation [13]. Aortic root dilatation in patients with QAV was reported in only 1 case [14]. Aortic valve regurgitation may induce diffuse dilatation of the ascending aorta. The opposite- aortic regurgitation secondary to aortic root dilatation - may also occur as a result from disruption or dissolution of elastic tissue within the aortic ring, as this structure provides the main support for the valve cusps [13].
CONCLUSION
It is worth noting that TTE could not provide the relevant AV anatomy. Both 2-dimentional and 3-dimentional TOE are first choice of investigation to make appropriate assessment of QAV. Prompt symptom recognition is pivotal and timed intervention can save patient’s life. Our case of type F QAV also provides a possible first link between hereditary aortic root disease and QAV.
ABBREVIATIONS
| AR | = Aortic regurgitation |
| LV | = Left ventricle |
| QAV | = Quadricuspid aortic valve |
| TTE | = Transthoracic echocardiogram |
| TOE | = Transoesophageal echocardiography |
CONFLICT OF INTEREST
All authors declare that they have no competing interests.
ACKNOWLEDGEMENTS
We are obliged to Mr Stefano Forlani (Cardiac Surgeon, Sheffield Teaching Hospitals NHS Foundation Trust) for providing us with the intra-operative picture of aortic valve.

