All published articles of this journal are available on ScienceDirect.

Niacin Modulates Pro-inflammatory Cytokine Secretion. A Potential Mechanism Involved in its Anti-atherosclerotic Effect
Authors Info & Affiliations
Abstract
The pathogenesis of atherosclerosis includes the assignment of a critical role to cells of the monocyte/macrophage lineage and to pro-inflammatory cytokines. Niacin is known to improve lipid metabolism and to produce beneficial modification of cardiovascular risk factors. The aim of this work was to investigate if Niacin is able to modulate pro-inflammatory cytokine production in macrophages in a murine model of atherosclerosis. For this purpose C57Bl/6J mice fed with atherogenic diet (AGD) or with conventional chow diet were used. The AGD group showed an increase in body weight and in total plasma cholesterol, with no differences in triglyceride or HDL levels. Lesions in arterial walls were observed. The characterization of Niacin receptor showed an increase in the receptor number of macrophages from the AGD group. Macrophages from control and AGD animals treated in vitro with an inflammatory stimulus showed elevated levels of IL-6, IL-1 and TNF-α, that were even higher in macrophages from AGD mice. Niacin was able to decrease the production of pro-inflammatory cytokines in stimulated macrophages. Similar effect of Niacin was observed in an in vivo model of inflammation. These results show an attenuating inflammatory mechanism for this therapeutic agent and would point out its potential action in plaque stabilization and in the prevention of atherosclerosis progression. Furthermore, the present results provide the basis for future studies on the potential contribution of Niacin to anti-inflammatory therapies.
INTRODUCTION
Atherosclerosis is a complex vascular disease that usually begins in the first decade of life and is now recognized as an inflammatory illness [1]. It is a process characterized by the accumulation of lipids, mononuclear cells, fibrous components, and calcium in the arteries [2]. It begins in early life and progresses through adulthood, exacerbated by hypertension, diabetes, hyperlipidemia, smoking, and obesity [2-4]. Vascular injury, recruitment of monocyte cells and infiltration of foam cells (lipid-laden macrophages) in combination with T lymphocytes, promotes the lesion formation [2,4]. The deposition of lipids is accompanied by migration and proliferation of smooth muscle cells.
Nicotinic acid, also known as Niacin, is well established for the treatment of dyslipidemia, as it has a potent effect in lowering plasma LDL-cholesterol and raising HDL-cholesterol. Numerous clinical studies have demonstrated a significant reduction in cardiac events and in cardiovascular disease-related mortality with Niacin treatment [5, 6]. However, the exact mechanism of action of this drug is still not fully understood. The orphan receptor GPR109A, a G-protein-coupled receptor (GPCR) was recently demonstrated to bind and to be activated by Niacin [7, 8]. This receptor has recently been given the HGNC approved gene symbol, NIACR1, but it is also termed HM74a in humans and PUMA-G (for protein up-regulated in macrophages by interferon-gamma) in mice [7, 8]. The discovery of a very highly expressed GPR109A that binds Niacin with high affinity in adipocytes has conducted the research of the antilipolytic effects of Niacin in adipose tissue [7, 8]. Acting through the GPR109A it was shown that Niacin suppresses the release of free fatty acids (FFA) from adipose tissue, and it is believed that the consequent reduction in free fatty acid flux to the liver leads to the decreased synthesis of triglyceride and of VLDL production by substrate depletion. In patients nicotinic acid reduces LDL-cholesterol and increases HDL cholesterol [9]. The observed reduction in the progression of atherosclerosis and in cardiovascular morbidity with Niacin would be solely due to its lipid modifying effects [5].
However, inflammation is a key component of atherosclerosis and genes coding for inflammatory or anti-inflammatory molecules are, therefore, good candidates for estimating the risk of developing atherosclerosis [10-12]. Accordingly, the production of high inflammatory molecules has been associated to atherosclerosis [13-15]. It is possible to postulate that drugs with a positive control of inflammation might play a protective role against atherosclerosis and would be potential therapeutic candidates to prevent atheroma formation.
A key question is whether the activity of nicotinic acid via GPR109A expressed on the surface of the adipocyte is sufficient to account for the overall clinical efficacy observed. In addition to its expression on adipocytes, GPR109A is known to be highly expressed also in spleen, lung, and lymphoid cells [8]. To date, there are studies suggesting GPR109A-mediated effects outside the adipose tissue. Experimental findings indicating high expression of GPR109A in lymphoid cells together with the observation of its up-regulation in murine macrophages by cytokines like IFN-γ [16], would suggest a role for GPR109A in immunity and inflammation. In recent years, the anti-inflammatory effects of Niacin have attracted attention, but the exact knowledge on the mechanisms involved in these actions is still missing. Additionally, some previous studies in vitro and in vivo have revealed that Niacin has anti-inflammatory properties that are independent of its effects on lipid regulation [17-21].
In this scenario, the aim of the present work was to investigate if Niacin actions involved the modulation of pro-inflammatory cytokine production in macrophages from mice submitted to an atherogenic diet, analyzing the involvement of macrophage Niacin receptor in these effects. To study experimental atherosclerosis we use C57BL/6J mice strain as it was demonstrated to be the most atherosclerosis sensitive strain [22].
MATERIALS AND METHODS
Mice
Inbred female C57Bl/6J mice, 4 weeks old, were obtained from the Facultad de Veterinaria, Universidad de Buenos Aires (UBA), Argentina and were housed in the animals facility of the Centro de Estudios Farmacológicos y Botánicos (CEFYBO)- Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET)- UBA. Animals were maintained on a 8:00 am to 8:00 pm light/dark cycle under controlled temperature (18-22ºC). All animal experiments and care were in accordance with the principles and guidelines of the Guide for the Care and Use of Laboratory Animals, US National Research (NIH) accredited by the Institutional Committee for use and care of Laboratory Animal (CICUAL, Facultad de Medicina, Universidad de Buenos Aires, Argentina). It is worth to note that this murine strain was previously shown to develop diet-induced fatty streak lesions [23].
Animal Model of Atherosclerosis
A total of 125 female mice were used for atherogenic diet and other 125 animals were assigned for the chow diet group. Animals were randomly assigned to two groups: one that was fed with conventional chow diet (Control mice) and the other with the atherogenic diet (AGD mice). This diet was a pellet obtained by mixing an special argentine commercially breeding chow containing 10% fat, 26 % proteins, 34 % carbohydrate, with the addition of 15% dairy butter. As previously described this type of diet exerts the largest atherogenic action [23,24]. The standard diet contains 26% proteins, 45 % carbohydrate and 5% fat. The two diets contain AIN-93 mineral and vitamin mix.
Histological Studies
Mice were anesthetized with 28 % (w/v) chloral hydrate, 0, 1 ml/ 100g of body, and perfused with 4% paraformaldehyde in phosphate buffer 0, 1 M, pH 7.4 through the abdominal aorta. Later this artery was dissected and the thoracic and abdominal segment selected and post fixed in the same solution during 2 hours, immersed overnight in phosphate buffer 0, 1 M, pH 7.4 and them embedded in paraffin for optical microscope evaluation. Sections of 4 µm were cut and mounted in silanized slices.
For immunohistochemical studies, artery sections (4 µm) were incubated overnight with the primary antibody (α- Actin, 1:100, monoclonal mouse antibody, 1A4, DAKO). Following several wash steps with PBS, artery sections were incubated with the proper secondary antibody (Vector Laboratories Inc., Burlingame, CA, USA) and followed by incubation with a biotin streptavidin complex (HRP Histo Mark, Caramillo, CA, USA). After washing in PBS, sections were developed and AEC substrate kit (Invitrogen Gaithersburg, MD, USA) until staining was optimal as examined by light microscopy using Leica Microscopes. Images were collected using a CCD video camera CU-M50 (Sony Inc.). Pictures were analyzed and compiled using Adobe Photoshop 8.0 CS3.
Purification of Murine Macrophages
Macrophages were purified from peritoneal exudates as previously described [25]. Briefly, after sacrifice, mice were injected intraperitoneally with 10 ml of sterile PBS and after 10 min the ascitic fluid was removed. This fluid was centrifuged 10 min at 200g and the cell pellet was washed twice with PBS. Cells were seeded at a concentration of 1x106cel/cm2 in culture medium (RPMI 1640, supplemented with 10% FCS and antibiotics) in 60x15mm Petri Dish with Cover (Corning®) to purify adherent cells. After 2 h non-adherent cells were discarded and macrophages were removed by trypsin treatment. This procedure renders approximately 2-4 x 106 macrophages per mouse.
Expression of GPR109A in Macrophages by [3H]-Nicotinic Acid Binding
Saturation binding assays were carried out on purified macrophages according to Lorenzen et al., 2001 [26] and
Wise et al., 2003 [8]. Briefly, 6 x 106 cells per binding point in a total volume of 250 μl were incubated with increasing concentrations of 5,6-3H nicotinic acid (American Radiolabeled Chemicals Inc, 50 Ci/mmol) ranging from 10 to 200 nmol/L for 3 h at room temperature with agitation. Nonspecific binding was assessed in the presence of 1 mM nicotinic acid. The bound ligand was recovered onto presoaked GF/B filters, washed four times with 1 ml of ice-cold binding buffer, and measured by liquid scintillation counting.
Cytokine Production
1 x 106 cells were incubated in 1 ml of supplemented medium (RPMI-1640 with 10% fetal calf serum and antibiotics) on 24 well-plates. These cells were incubated alone or with increasing concentrations of Niacin, ranging from 50 to 1000 μM and stimulated with lipopolysaccharide (LPS) and interferon-γ (IFN-γ) (Sigma Aldrich Co.) for 24 hours as previously described [27]. After incubation cell-free supernatants were obtained and the cytokine levels indicated in the Result section were measured by enzyme-linked immunosorbent assays (ELISA) using commercially available kits, following the manufacturer instructions. To further characterize the role of the GPR109A receptor, that is coupled to G protein, studies were also performed on macrophages pretreated with an inhibitor of G protein coupling, namely pertussis toxin (PTX), for 18 h.
Inflammation Mice Model
An additional group of mice (n=12) was used to study the effect of Niacin on in vivo cytokine production. For this purpose animals were divided in four experimental groups: injected intraperitoneally with PBS or 500 µg (20 mg/kg) of LPS (Escherichia coli 026:B6) and simultaneously oral treated or non-treated with Niacin (250 mg/Kg)). Twelve hour after the injection, intraperitoneal macrophages were obtained and incubated in supplemented medium for additional twelve hours to collect supernatants for cytokine analysis [28]
Statistical Analysis
Group differences were tested by one-way analysis of variance (ANOVA). When multiple comparisons were necessary after ANOVA, the Student-Newman-Keuls test was applied. Weight analysis was done with repeated measures ANOVA. When results showed not normally distribution the non-parametric Mann-Whitney test was performed. Differences between means were considered significant when p< 0.05.
RESULTS
Differences in Lipid Profile and Arterial Lesions between Control and AGD Mice
To validate the atherogenic response in AGD mice, serum lipid levels were measured in peripheral blood samples and tissue lesions were evaluated in aortic sections by immunohistochemistry. As shown in Fig. (1), AGD, but not control mice, displayed an increase in body weight and in cholesterol levels. No changes in triglycerides, glucose, HDL, LDL or VLDL were observed in both groups. Moreover these animals had important aortic lesions that were not present in control mice. As can be seen in Fig. (2), control animals showed aortic arteries sections without morphological changes. All layers were preserved and no evidence of cell migration or endothelial activation was observed (panel A). In AGD mice the arteries wall sections showed significant alterations such as inner elastic layer disruption, disorganization of smooth muscle cells (SMC) from middle layer, vacuolization and ulterior migration of smooth muscle cells to the neointimal layer and an aortic wall intimal thickening (panel B and E). Also, in control animal’s contractile cells (differenciated) predominate over the other group; opposite what was found in AGD animals (panel F). It is worth noting that myoepithelial cells were finally confirmed by their strong positive immunohistochemical staining with anti-(-actin antibody (Panel C). Moreover, the presence of CD68+ cells (macrophages) was observed (Panel D).
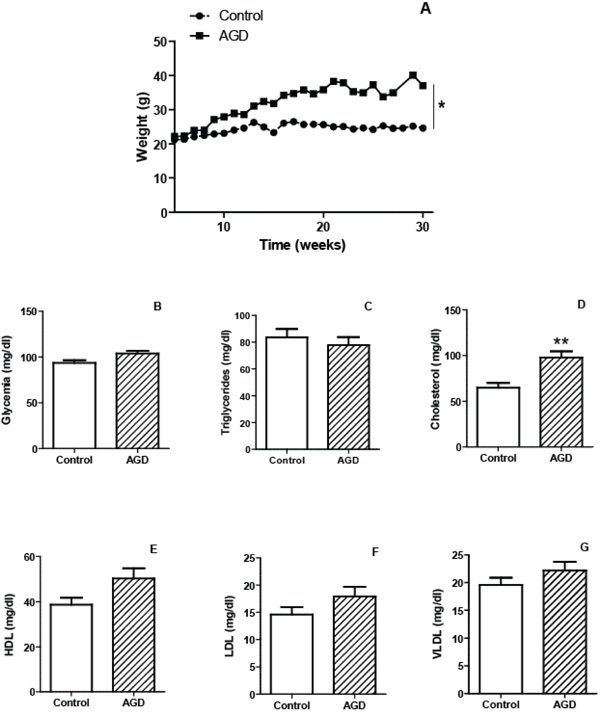
Characteristics of mice submitted to the atherogenic diet. The mean weights of n=30 control (circles) or 30 atherogenic diet-fed (AGD mice, squares) mice recorded at different weeks are depicted (panel A). Plasma levels of glycemia (panel B) and lipid profile: triglycerides (panel C) cholesterol (panel D), HDL (panel E), LDL (panel F) and VLDL (panel G) were determined for control and AGD mice. Results shown are the mean ± SEM of n = 30 mice from each group. Differs significantly from control animals, * p<0.05, ** p<0.01.
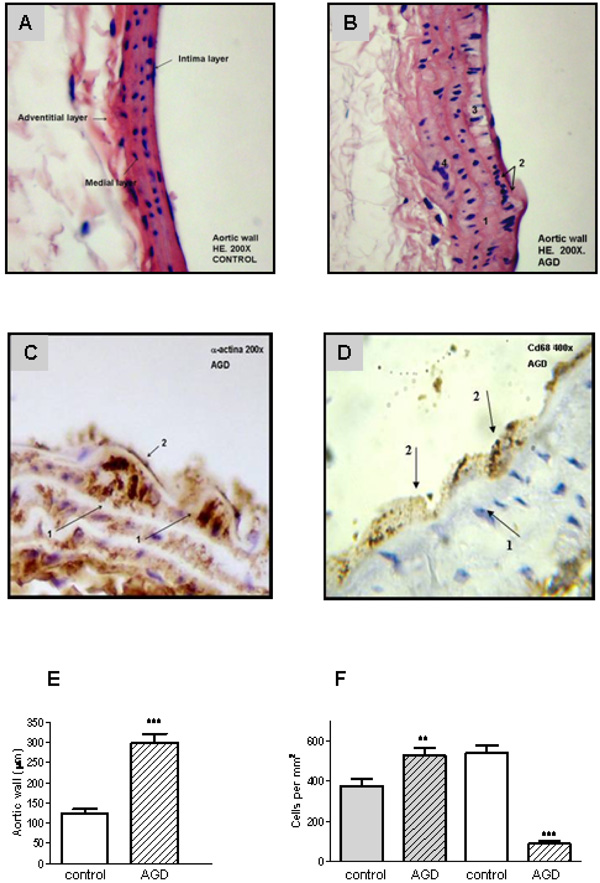
Aortic lesions in AGD mice. Histological sections of aortic arteries, representative of n=10 sections from different animals of each group, are shown. Panel A: Control aortic wall stained with Hematoxylin-eosin (HE) shows that all layers are conserved. Panel B: HE staining of aortic wall from AGD mice: 1. rupture of the internal elastic layer; 2. migration of smooth muscle cells into the neo-intima; 3. vacuolization of sub-intimal smooth muscle cells; 4. disruption of smooth muscle. Panel C: immunohistochemical staining with anti-a-actin antibody: 1-myoepithelial cells; 2-endothelial cells. Panel D: immunohistochemical staining anti-CD68 (macrophages). 1- myoepitelial cells, 2- macrophage. Panel E: size of the intimal aortic wall. Panel F: average number of medial smooth muscle cells (SMCs). This value of intimal cells was determined by counting nuclear profiles of two types of intimal cells: “synthetic”, undifferentiated cells (gray), and “contractile”, differenciated cells: (white). Results are expressed as cells per unit area of intima (i.e., cells per square millimeter) in control and treated (AGD) animals. Data represent mean ± SEM; n=3 in each group (Panel E and F), ** p< 0.01 and *** p<0.001
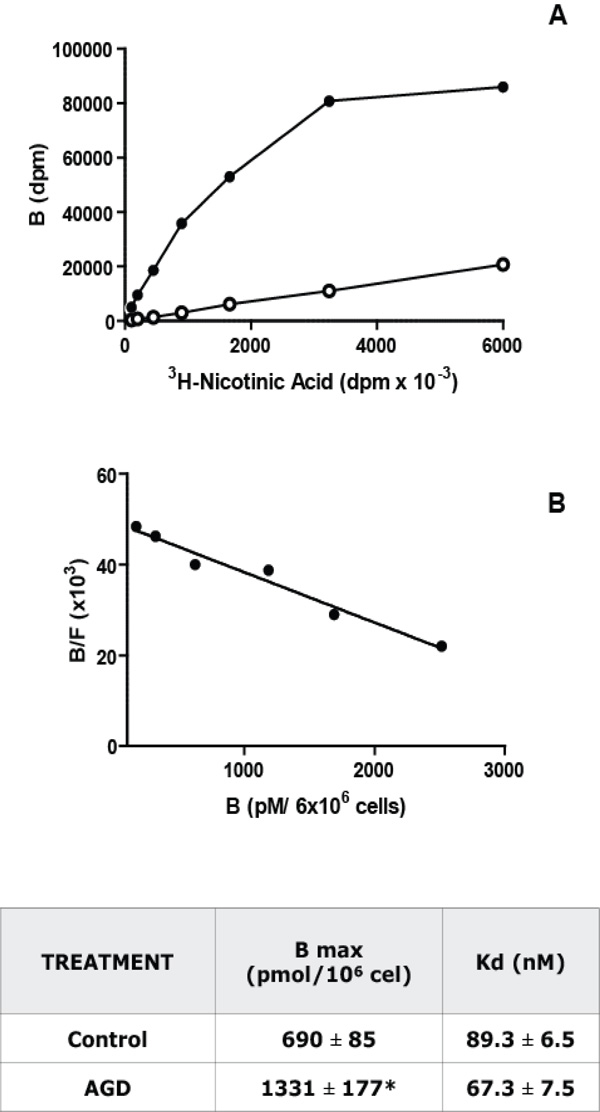
Niacin receptor characterization in macrophages. Panel A: Saturation Binding Assays: Increasing concentrations of [3H]- Nicotinic acid were incubated with 6 x 106 macrophages during 3 h at 25°C. Total (black) and non-specific (white, in the presence of 0.1 mM of Nicotinic acid) binding curves for [3H]-Nicotinic acid to isolated peritoneal macrophages from C57Bl/6J control mice were obtained. Panel B: Scatchard analysis of the results shown in panel A. The ratio of specific bound [3H]-Nicotinic acid to free (B/F) is plotted as a function of [3H]-Nicotinic acid bound (B). The intercept with the abscissa is the number of binding sites (Bmax) and the negative reciprocal of the slope is the dissociation constant (Kd). A representative experiment from 5 independent assays performed in duplicate is depicted in both panels. The Kd and Bmax values, mean ± S.E.M. from 5 experiments, for control and AGD mice macrophages are shown in the adjoined table. * p<0.05.
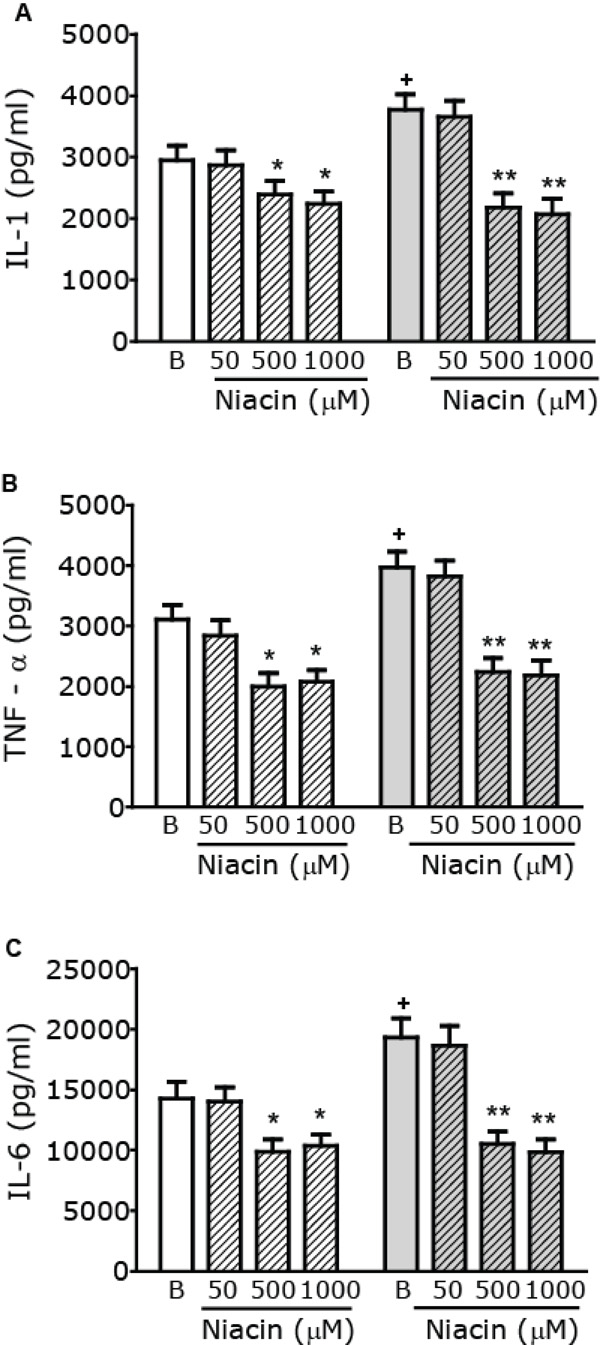
Modulation of macrophage pro-inflammatory cytokine production by Niacin. Macrophages from control (white) or AGD (gray) mice were incubated in the presence or absence of increasing concentrations of Niacin (µM) and stimulated with LPS (10 mg/ml) + IFN-γ (100 U/ml) for 24 h. Cytokine levels, namely IL-1 (Panel A), TNF-α (Panel B) and IL-6 (Panel C) were measured in cell free supernatants, using commercial kits. Results shown are the mean ± SEM of 3 independent experiments performed in duplicate. + p< 0.05 Basal (B) AGD vs Control mice; * p< 0.05, ** p<0.01 vs the corresponding B value.
Up-regulation of Nicotinic Acid Receptors in Macrophages from AGD Mice
To evaluate if the changes compatible with atherosclerosis induction in AGD mice were accompanied by modifications in macrophages, Niacin receptor expression was studied on purified macrophages from control and AGD mice by 3H Nicotinic acid binding. As can be seen in Fig. (3), a saturable and specific (more than 80%) binding of radiolabelled Niacin was obtained in these cells. Scatchard analysis of saturation curves showed that macrophages from AGD mice have twice binding sites than control mice. No differences were found in the nicotinic acid receptor affinity, as similar Kd were observed.
Niacin-mediated Modulation of Pro-inflammatory Cytokines
As basal macrophage cytokines levels in both control and AGD mice are too low, we used two experimental approaches to evaluate the effect of Niacin on cytokine production: i. by assaying in vitro cytokine secretion from macrophages after a pro-inflammatory stimulus and ii. by evaluating the production of cytokines in macrophages from mice injected with an inflammatory dose of LPS. To induce the production of pro-inflammatory cytokines, in vitro macrophages were stimulated with LPS and IFN-γ as described [27]. To study the action of Niacin on pro- inflammatory cytokine production, cells were stimulated in the absence or presence of increasing concentrations of Niacin (ranging from 50 µM to 1000 µM) and Tumor Necrosis Factor-α (TNFα), Interleukin-1 (IL-1) and Interleukin-6 (IL-6) levels were measured in cell free supernatants of these cultures. As shown in Fig. (4) stimulated cytokine production in the absence of niacin (Basal) is higher in macrophages from AGD than those from control mice. Pro-inflammatory cytokine production was reduced in the presence of 500 to 1000 μM Niacin both in control and AGD mice, being the effect more evident in AGD than in control animals. To further characterize the role of the GPR109A receptor, macrophages were pretreated with PTX, a nonselective G protein-uncoupling agent, frequently used to examine the function of G protein-coupled receptors. As can be seen in Table 1, pretreatment with PTX blocked the Niacin-mediated effect on pro-inflammatory cytokine production.
Effect of Pertussis Toxin (PTX) on Niacin (NA)-mediated Decrease of Stimulated Cytokine Secretion
| Cytokine | Mice Condition | Treatment | Cytokine Levels (pg/ml) |
|---|---|---|---|
| IL-1β | Normal AGD | LPS LPS+NA LPS+NA+PTX LPS LPS+NA LPS+NA+PTX |
3194 ± 303 2322 ± 267** 2995 ± 298# 3854 ± 321 2256 ± 287** 3764 ± 333# |
| TNF-α | Normal AGD |
LPS LPS+NA LPS+NA+PTX LPS LPS+NA LPS+NA+PTX |
3546 ± 294 2123 ± 189** 3721 ± 311# 4247 ± 395 2233 ± 227** 4323 ± 344# |
| IL-6 | Normal AGD |
LPS LPS+NA LPS+NA+PTX LPS LPS+NA LPS+NA+PTX |
15442 ± 1670 10456 ± 998** 16028 ± 1687# 21234 ± 1711 10786 ± 1213** 19999 ± 1532# |
Macrophages from control or AGD mice were pretreated with vehicle or PTX (50 ng/ml) for 18 h. After this time cells were incubated in the presence or absence of 1000 mM of Niacin (NA) and stimulated with LPS (10mg/ml) + IFN-g (100 U/ml) for 24 h. Cytokine levels were determined as described in Fig (4). Data are mean ± S.E.M. of three experiments in each group. **p<0.01 vs the corresponding basal value, # p<0.05 respect NA value.
Niacin Action on Cytokine Production by Macrophages from Mice Injected with LPS
| Treatment | Cytokines levels (pg/ml) | ||
|---|---|---|---|
| IL-1β | TNF-α | IL-6 | |
| PBS | Non detectable | 23 ± 14 | 35± 18 |
| LPS | 458 ± 57* | 856 ± 101* | 934 ± 123* |
| PBS + Niacin | Non detectable | 19 ± 16 | 32 ± 25 |
| LPS + Niacin | 209 ± 32* # | 486 ± 52* # | 492 ± 67* # |
Control mice were injected with PBS or LPS (20 mg/Kg) and were simultaneously orally treated with or without Niacin (250 mg/Kg). After 12 h, macrophages were collected and incubated for other 12 h in supplemented medium. Cell free supernatants were obtained to determine cytokine levels as described in Fig. (4). Data are mean ± S.E.M. of three animals *p<0.05 respect the corresponding PBS injected mice, #p<0.05 respect the corresponding non Niacin treated mice
To induce the production of pro-inflammatory cytokines in vivo, mice were injected with 500 μg of LPS. As shown in Table 2, production of IL-1, TNF-a and IL-6 for macrophages were significantly increased 12 h after LPS injection respect to PBS injected mice. Niacin treatment significantly reduced this elevated production of cytokines (Table 2).
DISCUSSION
Novel therapeutic opportunities could emerge from the knowledge of the role of inflammation in atherosclerosis. In this context, we here demonstrate that Niacin acts directly upon macrophages to reduce the release of pro-inflammatory cytokines. These findings would point out its potential action in the prevention of atherosclerosis progression and in plaque stabilization.
Many studies have explored the relationship between dietary factors, changes in lipoprotein levels, and risk for atherosclerosis, particularly, the regulation of cholesterol and lipoprotein metabolism by the type and amount of dietary fat. Therefore we chose a diet with saturated fatty acids from animal origin (dairy butter) in order to induce an aortic lesion area in normal C57Bl/6J mice. This diet contains a higher percentage of fat, with no cholic acid or cholesterol addition, and it mimics human’s intake of fat. After six months, animals feed with this diet showed only a significant increase in plasma cholesterol levels without significant changes in the lipoprotein profile. However, the arteries wall sections of these animals showed significant alterations such as inner elastic layer disruption, disorganization of smooth muscle cells (SMC) from middle layer and vacuolization and ulterior migration of smooth muscle cells to the neointimal layer. Also, undifferentiated cells predominate in AGD arteries. In fact, these cells are responsive to factors that direct migration and proliferation to the tunica intima and then distort the normal intimal architecture leading to the obstruction of flow and restenosis [29]. Many studies tried to unravel the role of SMC in atherosclerotic plaque formation. Initially the proliferation of these cells together with lipid infiltration of the intima was considered responsible for a slowly progressive process of plaque growth, eventually leading to lumen narrowing and ischemia [29]. By the other hand, SMC seem to protect the lesion against thrombosis, as they tend to encapsulate thrombogenic lipid together with macrophages and T-cells by means of forming a fibrous cap. Experimental models and analysis performed in human tissues determined that the SMC is the main cellular component in neointimal proliferation responsible for artery restenosis. Independently of the exact role that these SMC plays in arterial lesions, immunohistochemical findings observed in AGD mice are indicative of the induction of atheroma formation. According to these results, Lukasova et al [30] have recently reported aortic atherosclerosis lesions in a mouse model of atherosclerosis in the absence of HDL increment. Moreover, they also showed that nicotinic acid inhibited disease progression under conditions that left total cholesterol levels and cholesterol distribution among VLDL, LDL and HDL fractions unaffected. This highlights the fact that that nicotinic acid can reduce the progression of atherosclerosis independently of its lipid-modifying effects.
Atherosclerosis could be considered as a systemic inflammatory disease. So, peritoneal macrophages, considered as systemic markers of inflammation, were used to study Niacin receptor expression and the effect of Niacin in the secretion of pro-inflammatory cytokines. Our results indicate that peritoneal macrophages from AGD mice showed a two fold increase in Niacin receptor number. The possibility arises that this increase could be due to IFNγ secreted by activated T cells, as was previously described [16]. In support to this, macrophages from LPS-injected mice displayed a two-fold increase in Niacin receptor number than control animals (data not shown), as was previously described for PUMA-G mRNA levels in macrophages from microbial septic mice [16].
As cytokine production in non-stimulated animals is very difficult to determine, for the analysis of Niacin action upon pro-inflammatory cytokines we used both in vitro and in vivo experimental approaches for this purpose. Results showed that macrophages from both control and AGD mice respond to a pro-inflammatory stimulus in vitro with increasing secretion of TNF-α, IL-1 and IL-6, but this increment was significantly higher in AGD than in control mice. Moreover, Niacin addition was able to decrease pro-inflammatory cytokine secretion in both control and AGD mice, being the inhibitory effect more pronounced in AGD than in control animals (% inhibition for control and AGD mice, TNFα: 36 vs 44%; IL-6: 31 vs 46%; IL-1: 19 vs 42 %). These results are in accordance with recently published findings [31] showing that Niacin is able to reduce secretion of stimulated pro-inflammatory mediators in a human acute monocytic leukemia cell line. On the other hand, Niacin in vivo treatment was able to reduce pro-inflammatory cytokine production in macrophages from mice injected with LPS. Moreover, it was described that Niacin significantly inhibited TNFγ-induced monocyte adhesion to cultured human aortic endothelial cells and activation of nuclear factor-kappa B (NF-κB) [19, 20].
CONCLUSIONS
These results suggest that Niacin, in addition to be a lipid-modifying drug, has a beneficial action by attenuating inflammatory mechanisms involved in atherosclerosis pathology and plaque stabilization. Furthermore, the present results provide the basis for future studies on the potential contribution of Niacin to anti-inflammatory therapies.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
ACKNOWLEDGEMENTS
The authors thank Mrs María Rosa Gonzalez Murano for her excellent technical assistance, Daniel Gonzalez and Marcela Marquez for their invaluable help in the animal model and Patricia Fernández for secretarial assistance. This work was supported by a grant from Merck & Co. Inc. (PT000071011).

