All published articles of this journal are available on ScienceDirect.

Thrombospondin-1 in Early Flow-Related Remodeling of Mesenteric Arteries from Young Normotensive and Spontaneously Hypertensive Rats
Authors Info & Affiliations
Abstract
We tested the hypotheses that TSP-1 participates in the initiation of remodeling of small muscular arteries in response to altered blood flow and that the N-terminal domain of TSP-1 (hepI) can reverse the pathological inward remodeling of resistance arteries from SHR.
We measured (1) changes in gene/protein expression in MA of 6 week old WKY and SHR exposed to either increased (+ 100 %) or reduced blood flow (- 90 %) for 24-40 hours and (2) structural changes in MA of 12 week old SHR exposed for 3 days to hepI in organ culture.
In both HF and LF of WKY, mRNA expression of eNOS, sGCα1 and PKG1β were significantly reduced (p < 0.05), whereas mRNA of TSP1 was markedly increased (p < 0.05). In MA of young SHR, similar results were obtained except that eNOS mRNA was not reduced in LF. Expression of TSP1 protein was significantly increased in LF of young WKY and SHR (p < 0.05). Exposure of MA of 12 week old SHR to hepI (1 µmol/L) resulted in a rapid lumen diameter increase (+ 12 ± 2% after 3 days) without alteration in vascular reactivity, distensibility, media surface area or cell number.
These are the first observations of reduced gene expression of eNOS/sGC/PKG and increased expression of TSP1 at the initiation of arterial remodeling in young WKY and SHR, irrespective of its outward or inward outcome. Furthermore, a fragment of TSP-1 rapidly and directly reversed pathological inward arterial remodeling of SHR in vitro.
INTRODUCTION
In small muscular arteries of young normotensive rats, an increase in blood flow leads to an increase in lumen diameter that normalizes wall shear stress. The acute response consists of a reduction of the contractile activity of the vascular smooth muscle cells [1-3]. The response to a chronic increase in blood flow involves increases of the structural lumen diameter and of the arterial wall mass [4-7]. Flow-induced vasodilatation and remodeling are both endothelium-dependent [4, 8, 9]. In conditions of reduced blood flow, acute vasoconstriction and inward hypotrophic arterial remodeling are observed [6, 10]. There is strong evidence that an increase in shear stress can stimulate the activity and expression of NO-synthase in the endothelium [9, 11-13]. The roles of NO and its main receptor soluble guanylyl cyclase (sGC) in flow-related arterial remodeling are less clear.
We previously demonstrated dedifferentiation and increased turnover of smooth muscle cells (SMC) during flow-induced remodeling of small arteries [14]. This contrasts to the inhibitory effects of NO on SMC dedifferentiation and proliferation [15, 16]. In a RNA microarray analysis we observed that flow-related arterial structural changes were preceded by altered expression of a large number of genes involved in differentiation and proliferation of SMC [17]. This was in turn preceded by a rapid and transient change in the expression of a small number of genes including that for the 82 kDa subunit of sGC (sGCα1, downregulated) and that for thrombospondin-1 (TSP1, upregulated) [18]. The cGMP produced by sGC, stimulates protein kinase G (PKG), which is known to inhibit the expression of TSP1 in rat vascular smooth muscle cells [19]. A sequence from the N-terminal heparin-binding domain of TSP1 stimulates focal adhesion disassembly and increases cell migration through binding to a receptor co-complex of calreticulin-LRP1 [20-22]. These findings suggest that the eNOS/sGC/PKG system would be down-regulated at the transition from flow-induced vasodilatation to flow-induced remodeling, leading to increased TSP1 expression and matrix metalloproteinase 2 (MMP2) activation, and thereby stimulation of a de-adhesive state facilitating reorganization of the arterial wall material [10, 20, 23-25].
Flow-induced remodeling of small arteries is impaired by high blood pressure [26-28]. This might contribute to reduced collateralization and arteriogenesis in ischemia and to the increased vascular resistance that characterizes hyper-tension. There is convincing evidence in spontaneously hypertensive rats (SHR) and in human essential hyper-tension, that the extent of inward eutrophic remodeling of resistance arteries is of pathogenic significance [24, 29] and actually correlates with the incidence of life threatening events [30]. Hence, pharmacological interventions that stimulate outward remodeling could offer a therapeutic benefit.
The goal of this study was twofold. First, we tested the hypothesis that the eNOS/sGC/PKG pathway is down-regulated and that gene and protein expression of TSP1 are upregulated early (24-40 hours) after the induction of flow-related arterial remodeling in vivo. We used 6 week old WKY and 6 weeks old pre-hypertensive SHR rats to study differences in molecular and functional outcomes after alterations in blood flow. Secondly, we tested the hypothesis that a peptide mimetic of TSP1 (hepI) which induces cellular de-adhesion [19-22] can directly reverse the inward remodeling of resistance arteries of 12 week old SHR. For these purposes we used rat mesenteric small muscular arteries that were exposed to (1) altered blood flow in vivo or to (2) hepI during arterial organ culture at constant pressure and flow.
MATERIAL AND METHODS
Animals
Male normotensive Wistar-Kyoto rats (WKY) and Spontaneously Hypertensive rats (SHR) were obtained from Charles River (Maastricht, the Netherlands). All animals were housed separately and had free access to standard food (SRMA-1210; Hope Farms, Woerden, the Netherlands) and tap water. All experiments were conducted according to institutional guidelines. This investigation conforms to the Guide for Care and Use of Laboratory Animals as published by the US National Institutes of Health (NIH Publication No.85-23, revised 1996), and all experimental protocols were approved by the Animal Ethics Committee of the Maastricht University.
MA Ligation Model
Small mesenteric arteries (MA) of 6 week old WKY (n =27) and SHR (n =27) were exposed to altered blood flow in vivo as described previously [6, 14, 17, 31]. Briefly, rats were anesthetized with isoflurane (Abbott, Kent, UK) and after laparotomy, local blood flow was reduced (LF) by distal ligation of three alternate second order MA branches. The MA between these had compensatory enhanced flow (HF). We previously observed in WKY that the blood flow averages 10% and 200% in LF and HF respectively when compared to second order MA outside of the surgical area (normal flow, NF) and that these interventions ultimately result in inward hypotrophic (LF) and outward hypertrophic arterial remodeling (HF), respectively [6, 14, 31]. Animals received buprenorphine (0.05 mg/kg, s.c.; Schering-Plough, Utrecht, Netherlands) as an analgesic at three time-points: before surgery, at the end of the day of surgery and the next morning.
Gene Expression (RT-PCR)
At 24 hours after flow-modifying surgery, NF, HF and LF MA segments were isolated and pooled for each individual WKY and SHR rat (n = 9). To isolate RNA, Trizol reagent (Invitrogen, Leek, NL) in combination with RNeasy Mini Kit (Qiagen, Venlo, NL) was used. RNA (100 ng) was then used for first-strand cDNA synthesis with Ready-To-Go You-Prime First-Strand Beads (GE healthcare, Diegem, BE) and pd(N)6 random hexamer primer (GE healthcare, Diegem, BE). Plasmids containing gene fragments of rat MMP2, TSP1 and cyclophilin were used as standard curves, while a pool of cDNA of all samples was used for standard curves for the eNOS, sGCα1 and PKG1β genes. Taqman RT-PCR for MMP2, TSP1 and cyclophilin was performed in an ABI Prism 7700 SDS cycler (Applied-biosystems, Bleiswijk, NL) in combination with the appropriate fluorogenic probes, primer and qPCR MasterMix (Eurogentec, Maastricht, NL). Taqman RT-PCR for eNOS, sGCα1, PKG1β and cyclophilin was performed in a MyiQ iCycler (Biorad, Zwijndrecht, NL) with primers and qPCR MasterMix containing the DNA intercalating dye, SYBR-green. Each PCR reaction was performed in duplicate wells using the following conditions, 10 min at 95°C, followed by a total of 40 cycles of 15 s at 95°C and 1 min at 60°C (15 s at 95°C and 30 s at 58°C for SYBR-green). Gene expression levels were normalized to the house-keeping gene cyclophilin in each sample.
Contractile and Relaxing Reactivity
To evaluate consequences of altered gene expression for vasomotor control, 2 mm arterial segments were harvested at 24 (n=4) and at 32 hours (n=8) after flow modifying surgery in 6 week old WKY and SHR. Preparations were mounted in a 5 ml organ chamber (Danish Myotechnology, Aarhus, DK) filled with Krebs Ringer bicarbonate solution (KRB; 37°C; 95% O2 / 5% CO2) between an isometric force transducer and a displacement device. Their diameter was set at 90% of that of the resting vessel at a transmural pressure of 100 mmHg. Effects of the endothelium-dependent vasodilator acetylcholine, the NO-donor Na-nitroprusside and of the non-selective NOS-inhibitor Nω-L-nitro arginine methyl ester (L-NAME) were evaluated in the presence of 3 μmol/L indomethacin during contractions induced by an equiosmotic KRB solution containing 40 mmol/L K+. These steps were taken to rule out influences of prostaglandins and endothelium-derived hyperpolarizing factors. Because depolarizing high K+ buffer can stimulate release of neurotransmitters from peri-arterial nerves, experiments were initiated after exposure of the arteries to phenoxybenzamine (1 μmol/L, 10 min) and capsaicin (1 μmol/L, 20 min) which irreversibly block α-adrenoceptors and peri-arterial sensorimotor nerves, respectively [32].
Protein Expression
Protein levels were investigated in view of discrepancies between mRNA levels on the one hand and vasomotor responses on the other hand. At 32 hours and at 40 hours after flow-modifying surgery, NF, HF and LF MA segments (n=9, each group) were isolated and pooled from 3 WKY and 3 SHR rats and snap-frozen in liquid nitrogen. Pooled arteries were crunched at -80°C and resuspended in ice cold RIPA buffer (50 mM HCl pH 7.4, 150 mM NaCl, 1% NP-40, 0.25% Na-deoxycholate and 1 mM EDTA) containing protease and phosphatase-inhibitors (1mM PMSF, 1 mM NaVO4, 1 mM NaF, 1 µg/ml aprotinin, 1 µg/ml pepstatin and 1 µg/ml leupeptin) to extract total protein. The lysates were cleared by centrifugation. Protein content of the lysates was determined with the BCA method [33]. Lysates were subsequently run on a criterion bis/tris gradient gel, and blotted to a nitrocellulose membrane. Membranes were blocked with 1:1 oddysey blocking buffer:PBS and were subsequently incubated with antibodies diluted in 1:1 oddysey blocking buffer:PBS against: eNOS (BD Transduction Laboratories, Franklin Lakes, USA, 1:2500), sGCα1 (Santa Cruz, Santa Cruz, USA 1:2000), PKG1β (Biovision, Zurich, CH, 1:500), MMP2 (Santa Cruz, Santa Cruz, USA 1:500,detecting the pro (72kDa)- and the active (62kDa) form of MMP2) and TSP-1 (Abcam, Cambridge, USA, 1:500). Primary antibodies were detected with secondary antibodies labeled with the near infrared dye IRdye800 and visualized with an odyssey detection system. TSP-1 antibody was detected with secondary antibodies labeled with biotin. Biotin was subsequently detected with IRdye800 labeled streptavidin. Protein expression levels were normalized to the house-keeping protein GAPDH in each run.
Arterial Organ Culture
To investigate whether hepI could reverse inward arterial remodeling in hypertension, we used MA segments of 12 week old SHR in organ culture as described by Bakker et al. [10, 34]. The vessels were mounted on two glass micropipettes in a stainless steel organ chamber. Vessels were pressurized at 80 mmHg (no flow) and incubated in Hanks balanced salt solution (HBSS) for 60 min at 37°C, before baseline measurements of diameter and reactivity were obtained. Thereafter the vessels were exposed intra-luminally as well as extra-luminally to culture medium (Leibovitz (n=6) or 5% dFCS-DMEM (n=6)) with or without 1 μmol/L hepI for 3 days. The parallel experiments with Leibovitz and 5% dFCS in DMEM yielded quantitatively identical results. The data were pooled for subsequent analysis.
At the start of the experiments and at daily intervals, passive pressure-diameter (P/D) curves were constructed [13]. The culture medium was replaced by Ca2+-free HBSS containing 10 μmol/L Na-nitroprusside to inactivate arterial smooth muscle tone. The cannulated arterial segments were pressurized using a feedback controlled pressure source (Living Systems Instrumentation, Burlington, USA) and placed on the stage of an invertoscope (Nikon TMS, Abingdon, USA) equipped with a video camera (Stemmer) and a digital device (LSI) for recording of lumen diameter. Intra-arterial pressure was increased in 10 mmHg steps from 20 to 120 mmHg. After each step, the vessel was allowed to equilibrate until a stable diameter was reached (2 minutes). Findings were expressed as percentage change from vessel diameter on day 0 at 80 mmHg. From the P/D relationships arterial distensibility was calculated by the following formula: DC = (1/ΔP)*(ΔD/Dn-1)*100 (DC = distensibility, P = intraluminal pressure, D = diameter at a given intraluminal pressure). After the highest pressure was reached, pressure was returned to 80 mmHg and the HBSS-solution was replaced by culture medium with or without 1.0 μmol/L hepI.
On day 0 and day 3 of the culture period the viability of the vessels was verified by recording diameter changes at 80 mmHg in response to 1 μmol/L serotonin, 10 μmol/L phenylephrine and 1 μmol/L acetylcholine in HBSS. Finally the vessels were fixed in neutral buffered formalin at 80 mmHg and 37°C and stored in 70% ethanol.
Histology and Morphometry
Fixed vessels were embedded in paraffin and cross-sections (4 μm) were stained with Lawson’s solution (Boom, Meppel, the Netherlands) to visualize the internal and external elastic laminae. Cross-sectional area (mCSA), thickness, cellular density of the tunica media, and the lumen diameter were determined as previously described [6, 13, 14, 17, 26, 31].
Solutions, Culture Media and Drugs
Leibovitz medium (Invitrogen, Leek, NL) was supplemented with 1 mg/mL albumine (Sigma Aldrich, Zwijndrecht, NL), 20 mg/mL ciprofloxacin (Bayer, Leverkusen, DE), 100 U/mL penicillin, 100 μg/mL streptomycin and 2.5 U/mL amphotericin B (Invitrogen, Leek, NL). DMEM (Invitrogen, Leek, NL) was supplemented with 2.5 mM glutamine, 100 IU/mL penicillin, 100 μg/mL streptomycin and 5% dialyzed fetal-calf serum. All pharmacological agents were purchased from Sigma Aldrich, (Zwijndrecht, NL). The hepI peptide (ELTGAARKGSGRRLVKGPD) was synthesized by Anaspec Inc (San Jose, USA) and purified by HPLC [21]. The peptide was confirmed to be >95% pure by HPLC and mass spectrometry.
Statistical Analysis
Results are expressed as means ± SEM. Statistical significance of differences between groups was evaluated by analysis of variance (ANOVA for consecutive measurements for pressure-diameter curves) or 1-way ANOVA followed by Bonferroni or paired t-test (SPSS 11.5). A value of P<0.05 was considered significant.
RESULTS
Gene Expression Under Basal Conditions
Fig. (1) illustrates expression of genes encoding protein kinase G (PKG) and candidate modulators and targets of this enzyme in mesenteric small arteries of 6 week old WKY and SHR rats. In control arteries with normal flow (NF), mRNA levels were high for sGCα1 and PKG1β, intermediate for eNOS, and low for MMP2 and TSP1. Although expression of eNOS tended to be smaller in NF of SHR, the expression levels of all five genes did not differ significantly between NF arteries of 6 weeks old SHR and WKY rats.
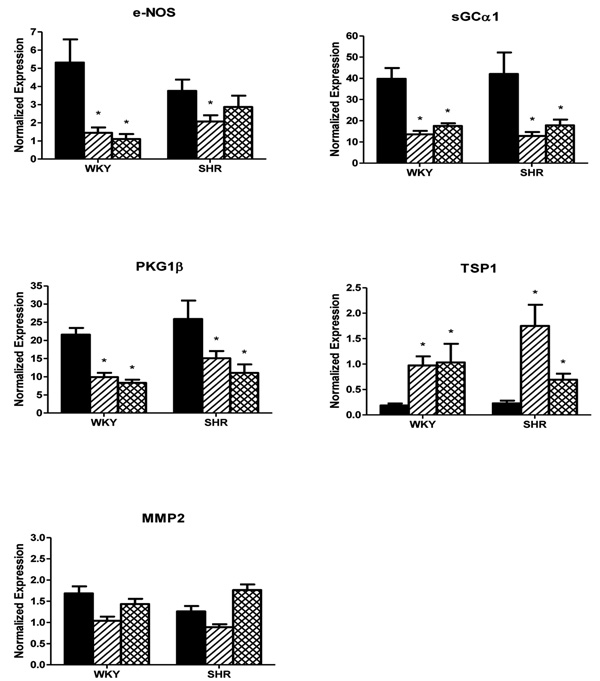
mRNA expressions of eNOS, sGCα1, PKG1β, TSP1 and MMP2 in mesenteric small arteries exposed to normal blood flow (NF, black bars) or for 24 hours to increased blood flow (HF, hatched bars) or reduced blood flow (LF, crosshatched bars) in 6 week old Wistar Kyoto (WKY) rats and Spontaneously Hypertensive rats (SHR) in vivo, normalized to the housekeeping gene cyclophylin. Results are expressed as the mean ± SEM (n = 6). * p<0.05 versus NF.

In vitro contractile reactivity of mesenteric small arteries of 6 week old WKY rats (panel A) and SHR rats (panel B) exposed to normal flow (black bars) or for 32 hours to increased (hatched bars) or reduced blood flow (cross-hatched bars). Means ± SEM (n = 8-11). Normalized diameter and maximal contraction in response to 125 mmol/L K+ (top left) and the effect of indomethacin and L-NAME on contractile responses to 40 mmol/L K+ (top right) are shown along with the relaxing responses to acetylcholine during K+-induced contraction in the presence of indomethacin (bottom left) and the relaxing responses to Na-nitroprusside during K+-induced contraction in the presence of indomethacin and L-NAME (bottom right) for NF (closed circle), HF (open circle) and LF (closed triangle). *p<0.05 versus NF; #p<0.05 INDO versus INDO + L-NAME.
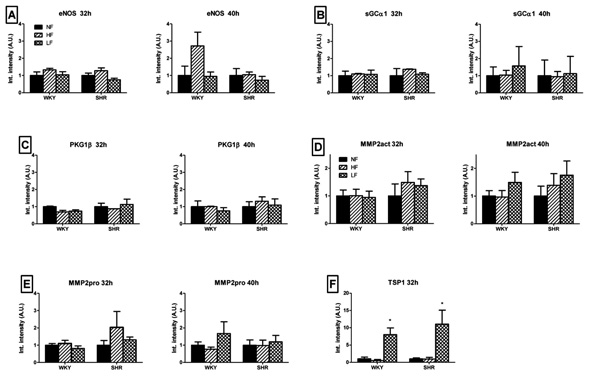
Changes in protein expression of eNOS (A), sGCα1 (B), PKG1β (C), MMP2 (pro/act, D/E) and TSP1 (F), in mesenteric small arteries exposed for 32 and 40 hours to normal (black bars), increased (hatched bars) or reduced (crosshatched bars) blood flow in 6 week old WKY and SHR rats in vivo. Means ± SEM (n = 6). *p<0.05 versus NF.
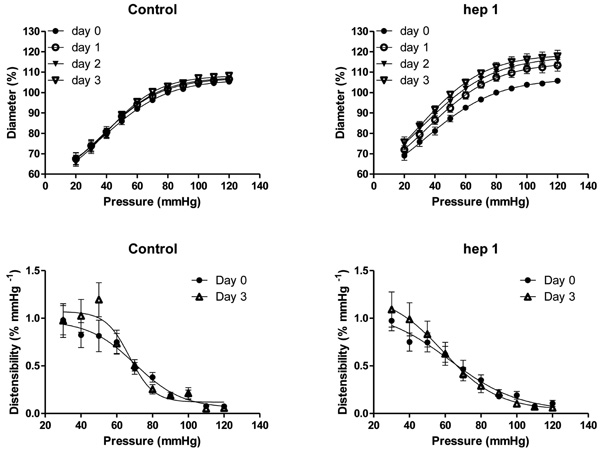
Effects of time (control, top left) and 1 µmol/L hepI (hepI, top right) on passive pressure-diameter relationships in mesenteric small arteries (MA) of 12 week old SHR during organ culture. Calculated distensibility for control MA (day 0 and day 3, bottom left) and hepI treated MA (day 0 and day 3, bottom right). Values are shown as means ± SEM (n = 12). Closed circle, day 0; open circle, day 1; closed triangle, day 2; open triangle, day 3. *p<0.05, versus day 0.
Altered Gene Expression During Initiation of Flow-Related Arterial Remodeling In Vivo
Changes in gene expression in MA exposed for 24 hours to altered blood flow in vivo are summarized in Fig. (1). Consistent with earlier RNA microarray observations [17, 18], the expression of sGCα1 was significantly reduced and that of TSP1 was markedly increased in arteries from WKY rats exposed to high blood flow (HF). Surprisingly, the findings were very similar in vessels exposed to increased (HF) or reduced (LF) blood flow. In both HF and LF arteries of WKY rats, the expression of eNOS, sGCα1 and PKG1β was significantly reduced, whereas TSP1 was markedly increased and MMP2 was not appreciably altered. In HF of SHR, qualitatively and quantitatively similar findings were obtained, except that there was a less marked downregulation of eNOS possibly due to the already reduced basal levels. Also in arteries of SHR exposed to reduced blood flow (LF), largely similar results were obtained, with the exception that eNOS and TSP1 expression were affected to a lesser extent. Collectively these results indicate that either an increase or a decrease in blood flow both lead to a concurrent reduction of eNOS, sGCα1 and PKG1β and to a marked increase of TSP1 mRNA levels within 24 hours in rat mesenteric small muscular arteries.
Vasomotor Responses
To evaluate potential functional consequences of the observed downregulation of the NO/sGC/PKG pathway, we recorded arterial contractile reactivity in vitro in MA of 6 week old WKY and SHR rats. At 24 hours after flow-modifying surgery there were no statistically significant differences between NF, HF or LF arteries (n = 4-6, data not shown). Because changes in the abundance and activity of proteins lag behind changes in mRNA expression, functional analyses were repeated at 32 hours after altered blood flow in vivo. These results are summarized in Fig. (2). The diameter of WKY LF arteries was significantly reduced which is consistent with the rapid inward remodeling of LF in vivo [14]. L-NAME significantly increased the contractile response to 40 mmol/L K+ in NF arteries of WKY and SHR. Relaxing responses of depolarized vessels to acetylcholine (in the presence of indomethacin) and to Na-nitroprusside (in the presence of indomethacin and L-NAME) did not differ between NF, HF or LF arteries (Fig. 2A). These findings indicate that the marked and concurrent reductions of the mRNA levels for eNOS, sGCα1 and PKG1β were not accompanied by reduced relaxing responses to basal and stimulated endothelium-derived or exogenous NO during initiation of outward (HF) or inward (LF) flow-related arterial remodeling in WKY. Similar results were obtained in SHR, as shown in Fig. (2B). In contrast to WKY, the diameter of SHR LF arteries did not differ from that of NF and HF arteries.
Altered Protein Expression During Initiation of Flow-Related Arterial Remodeling In Vivo
Fig. (3) summarizes changes in protein expression in MA exposed for 32 (left panel) and 40 hours (right panel) to altered blood flow in vivo. These time-points were chosen to allow protein synthesis to occur and are based on the 24 hour gene expression experiments and the earlier observation that in low flow conditions specifically, the inward remodeling response manifests within 2 days [14]. In both HF and LF arteries of WKY and SHR rats, the protein levels of eNOS, sGCα1, PKG1β and MMP2 (pro/act) were not significantly altered after 32 and 40 hours of flow modifying surgery (Fig. 3A-E). Protein expression for TSP1 was significantly increased in LF arteries of both WKY and SHR (Fig. 3F). After 40 hours of exposure to LF, protein expression of TSP1 was below threshold in NF, HF and LF arteries.
Arterial Structural Effects of the hepI Sequence of TSP1
To evaluate structural consequences of upregulated TSP1 mRNA and protein expression, isolated arteries were exposed to hepI under constant conditions of pressure (80 mmHg) and flow (no flow) in vitro. These experiments were deliberately performed in arteries of 12 week old SHR that display an inward eutrophic remodeling which contributes to the increased peripheral resistance and blood pressure [24, 29]. In the untreated arteries, the diameter at 80 mmHg and the relationship between passive diameter and transmural pressure did not change during the 3 day culture period (Fig. 4). The presence of 1 μmol/L hepI, however, resulted in an increase of the diameter at 80 mmHg and in an upward shift of the pressure-diameter curves (p < 0.05 for day 1, day 2 and day 3)(Fig. 4). Arterial structural diameter was increased by 7.5 ± 1.9 % within 24 hours and increased further to +12.6 ± 2.4 % after 3 days (Fig. 4). Exposure to hepI did not modify calculated distensibility (Fig. 4), general arterial histology (not shown), and did not result in statistically significant changes in media cross sectional area (13693 ± 4043 μm2 versus 12881 ± 3182 μm2) or cell number (52 ± 11 versus 54 ± 8 nuclear profiles/section) indicating that the hepI-induced outward remodeling was eutrophic in nature.
In an additional set of experiments we tested whether hepI displayed vasomotor effects. Contractile responses of freshly isolated arteries to 25 mmol/L K+ were reduced by acetylcholine and enhanced by indomethacin and L-NAME indicating intact endothelium. Depolarization-induced contractions were not modified by 0.0001 - 1.0 μmol/L hepI indicating that hepI does not display major acute vasomotor effects. After 3 days in vitro, 1 μmol/L serotonin and 10 μmol/L phenylephrine reduced arterial diameter to 45 ± 4% and 47 ± 3% of baseline, respectively, in untreated vessels and to 44 ± 3% and 46 ± 4% of baseline, respectively, in arteries that had been exposed to hepI. Moreover, 1 μmol/L acetylcholine reversed the serotonin-induced constriction by 66 ± 5% in untreated vessels and by 63 ± 5% in hepI treated arteries. These findings indicate that exposure to hepI for three days did not adversely affect vasoconstriction or endothelium-dependent vasodilatation.
DISCUSSION
We demonstrate for the first time i) rapid and concurrent downregulation of eNOS, sGC and PKG mRNA and upregulation of TSP1 mRNA expressions at the initiation of flow-related remodeling and ii) a direct effect of the calreticulin/LRP1 binding sequence of TSP1 on the structural lumen diameter in small muscular arteries of the rat. That the latter was observed in the narrowed arteries of SHR rats, suggests a therapeutic potential for the pathways involved.
In arteries, wall shear stress and circumferential wall stress are homeostatically controlled by vasomotor and structural changes of lumen diameter and wall thickness [6, 9, 35]. The transition from the acute vasomotor responses to the chronic structural responses is poorly understood. Here we concentrated on subacute effects of altered local blood flow in the interconnected arterial trees of the mesenteric vascular bed. In this system of young rats, increased and reduced blood flow ultimately lead to outward hypertrophic and inward hypotrophic remodeling, respectively [5-7, 14, 17, 18, 27, 28, 31]. Both structural responses involve marked changes in gene expression and in cellular turnover and differentiation [14, 17] which are preceded by a rapid and transient transcriptional response of a comparatively small number of genes [18]. The latter candidate initiators of remodeling might be a target for interventions aimed at improved collateralization in ischemia and reversal of the adverse structural narrowing of resistance arteries in hypertension. We focussed on changes occurring within 24 hours in a cell-cell signaling and signal transduction system that was shown to be involved in flow-induced vasodilatation [9, 11-13], vascular smooth muscle cell de-differentiation and proliferation [15, 16] and in the expression of a modulator of cellular adhesion and extracellular matrix, such as TSP1 [19, 22].
The wall shear stress resulting from blood flow can alter the activity and expression of type III NOS (eNOS) in the endothelium [9, 11, 12]. The main NO-receptor in vascular smooth muscle is the heterodimeric soluble guanylyl cyclase sGCα1β1 [36-38]. The cGMP produced by this enzyme stimulates protein kinases, in the vascular wall primarily PKG1β [36-38], which inhibit vasoconstriction, dedifferentiation and expression of TSP1 in vascular smooth muscle [19, 22]. In this study we confirmed several earlier findings like (i) inhibitory effects of low blood flow on the mRNA expression of eNOS [9, 12] and (ii) association between reduced mRNA expression of PKG and increased mRNA expression of TSP1 [19, 22]. The overall picture of our observations is, however, unexpected. Reduced mRNA expression of eNOS was accompanied by reduced mRNA expression of sGCα1 and PKG1β, which is not observed in eNOS-deficient mice [36, 37, 39, 40]. The mRNA expression of both sGCα1 and of PKG1β was reduced, although in a variety of vascular SMC culture models a negative feedback interaction between these effectors has been demonstrated [41]. Most surprising is that concurrent down regulation of eNOS, sGCα1 and PKG1β gene expressions were observed in vessels exposed to reduced blood flow and in vessels exposed to increased blood flow. This was found in two strains of rats (WKY and SHR). The finding suggests a general response to an altered hemodynamic micro-environment that prepares the tissue for adequate structural changes aimed at restoring stresses independent from the direction of the initiating change. This may be required to initiate cellular de-differentiation, de-adhesion and turnover and extracellular matrix reorganization that take part in both outward hypertrophic remodeling and in inward hypotrophic remodeling.
In addition, we studied protein expression and vasomotor responses to monitor turnover and activity of eNOS/sGC/PKG pathway components in response to altered blood flow in vivo. Protein expression of TSP-1 was increased in LF MA of both WKY and SHR at 32 hours after flow modifying surgery. However, no statistically significant changes in the protein levels of the other proteins and in vasomotor responses to NO-synthase blockade, an endothelium dependent vasodilator and an NO-donor were observed after 32 or 40 hours exposure of rat arteries to altered blood flow.
Although shear stress can at least transiently stimulate activity and expression of eNOS [9, 11, 12], there is no consensus on the precise role of eNOS in flow-induced arterial remodeling. Caveolin-mediated activation of eNOS seems to be required for the inward remodeling of ligated carotid arteries in mice [42]. In a rabbit carotid artery A-V fistula model, chronic L-NAME treatment inhibits outward remodeling and the contribution of NO can be attributed to stimulation of MMP9 [43]. Chronic L-NAME does, however, not impair inward and outward remodeling of mesenteric small arteries in the rat model that we used here [31]. In this rat mesenteric model, doubling of blood flow does not result in increased expression of eNOS after 2 or 7 days, whereas a four fold increase in blood flow leads to a significant increase in eNOS at these time points [44]. In a similar mouse mesenteric model, Dumont et al. [7] observed increased expression of eNOS and activity of MMP9 at 4 days that contributed to the structural outcome. Differences between species, arteries and extent of blood flow alterations may contribute to these divergent findings. TSP1, as already mentioned, is a large matricellular protein with many functions, particularly the ability of the hepI sequence of TSP1 to signal cellular de-adhesion [22]. This sequence of TSP1 binds to the receptor co-complex of calreticulin/LRP1 and stimulates focal adhesion disassembly and increased cell migration [20, 22, 45, 46]. To evaluate a possible contribution of TSP1 to small artery remodeling arteries were exposed to the hepI peptide mimetic in vitro [19].
As described by Bakker et al [34, 47], rat mesenteric small arteries could be maintained for 72 hours in arterial organ culture at constant pressure and in the absence of flow without noticeable alterations in viability of the smooth muscle and endothelium. In this static in vitro setting the arterial diameter did not change while narrowing of LF arteries developed within 40 hours in vivo (present study and [14]). This indicates that the absence of flow is in itself not a potent stimulus for inward remodeling. Administration of hepI in vitro resulted in a diameter increase and in an upward shift of the pressure-diameter relationship within 24 hours. The 12% increase in lumen diameter observed with hepI in vitro is comparable to the diameter response to doubling of blood flow in vivo [14, 17, 31]. HepI-induced outward remodeling was not accompanied by changes in wall mass or vasomotor control. Although TSP1 was upregulated in vessels exposed to increased or reduced blood flow in vivo, exogenous hepI caused outward, but not inward arterial remodeling. The role of vasomotor responses in arterial remodeling may help explain this paradox. Prolonged vasoconstriction has been shown to reorganize cell-matrix interactions [23] and to lead to a narrower arterial lumen (inward remodeling) in vitro [10, 48]. We now propose that TSP1 expressed in response to a change in shear stress can lead to an intermediate de-adhesive state [20] and that the levels of transmural pressure and vasoconstrictor/vasodilator tone will determine the outward or inward nature of the resulting arterial structural change. A distending pressure, but no vasomotor tone was present during culture allowing the hepI induced de-adhesive state to result in outward arterial remodeling. That 3 days of exposure to hepI increased diameter without altering arterial distensibility contributes to the suggestion that the remodeling resulted from a de-adhesive state rather than from major changes in the extracellular matrices.
Effects of altered blood flow on the mRNA’s and proteins investigated were largely similar in 6 week old WKY and SHR. This suggests that the pathways investigated are not likely involved in the inward remodeling of resistance arteries that contributes to the elevated total peripheral vascular resistance and blood pressure in SHR [24, 29, 30]. Despite this limited pathophysiological relevance, hepI, that lacks apparent vasomotor effects was found to rapidly and directly reverse inward arterial remodeling of mesenteric resistance arteries of the hypertensive 12 week old SHR. This strengthens the proposal that small artery structure is a directly amenable pharmaco-therapeutic target.
CONFLICT OF INTEREST
Declared none.
ACKNOWLEDGEMENTS
Parts of this work were financed by the European Vascular Genomics Network (http://www.evgn.org) supported by the European Community’s sixth Framework Programme (contract LSHM-CT-2003-503254), a travel grant from the Italian Society for Hypertension to GB and by a grant from the Dutch Top Institute Pharma (T2-108; metalloproteases and novel targets in endothelial dysfunction).

