All published articles of this journal are available on ScienceDirect.

Cardiac Ischemia and Ischemia/Reperfusion Cause Wide Proteolysis of the Coronary Endothelial Luminal Membrane: Possible Dysfunctions
Authors Info & Affiliations
Abstract
Background:
Ischemia and ischemia-reperfusion (I/R) are common clinical insults that disrupt the molecular structure of coronary vascular endothelial luminal membrane (VELM) that result in diverse microvasculature dysfunctions. However, the knowledge of the associated biochemical changes is meager. We hypothesized that ischemia and I/R-induced structural and functional VELM alterations result from biochemical changes. First, these changes need to be described and later the mechanisms behind be identified.
Methods:
During control conditions, in isolated perfused rat hearts VELM proteins were labeled with biotin. The groups of hearts were: control (C), no flow ischemia (I; 25 min), and I/R (I; 25 min, reperfusion 30 min). The biotinylated luminal endothelial membrane proteins in these three different groups were examined by 2-D electrophoresis and identified. But, it must be kept in mind the proteins were biotin-labeled during control.
Results:
A comparative analysis of the protein profiles under the 3 conditions following 2D gel electrophoresis showed differences in the molecular weight distribution such that MWC > MWI > MWI/R. Similar analysis for isoelectric points (pHi) showed a shift toward more acidic pHi under ischemic conditions. Of 100 % proteins identified during control 66% and 88% changed their MW-pHi during ischemia and I/R respectively. Among these lost proteins there were 9 proteins identified as adhesins and G-protein coupled receptors.
General significance:
I and I/R insults alter MW-pHi of most luminal glycocalyx proteins due to the activation of nonspecific hydrolizing mechanisms; suspect metalloproteases and glycanases. This makes necessary the identification of hydrolyzing enzymes reponsible of multiple microvascular dysfunctions in order to maintain the integrity of vascular endothelial membrane. VELM must become a target of future therapeutics.
1. INTRODUCTION
Clinically myocardial ischemia and ischemia/reperfusion (I/R) insults; a scientific and a major clinical problem [1], indiscriminatingly occur during cardiac transplant, coronary bypass, angioplasties, thrombolysis, open heart surgery and all interventions that result in transient interruption of an organ circulation. All these beneficial procedures have brought relieve to millions of patients, but, the associated I/R insult causes diverse cardiac dysfunctions whose mechanisms, by not being well understood prevent correct treatments. I/R injure different cell types and their organelles [2-8]. However, the coronary vascular endothelial luminal membrane (VELM) despite its role on infarct size [10, 12, 13],it is suspect I/R induce important chemical structural changes and as compared to these changes in other organelles [2-5, 8, 9], VELM changes are unknown.
VELM or ”luminal endothelial glycocalyx”, is a molecular complex composed of a large variety of extracellular domains of transmembrane proteins, glycoproteins, proteoglycans, glycosaminoglycans and matrix bound plasma proteins [13-16], consequently in the VELM reside multiple normal and pathological functions; solute wall permeation, flow sensing, hormone reception, coagulation, atherosclerosis etc [7,12,13,17-19, 21-23]. All these endothelial luminal functions are disturbed by ischemia and I/R and associated oxygen radicals [1, 20, 24]. The luminal endothelial glycocalyx has a thickness estimated between 0.2 - 0.8 μm [11-14, 18, 20]. In living microcirculatory preparations in which fluorescently labeled dextran of various sizes (50 to 2,000 kD) were infused intraarterially. Fluorescence distributed uniformly in most of the lumen of blood vessels, but was absent in a zone 0.2 – 0.8 μm thick adjacent to the vessel wall, an “endothelial exclusion zone” (EEZ). Infusion of dextrans of lower molecular weight resulted in a deeper penetration implying that EEZ constitutes a barrier for solute difussion likely the result of a restricted diffusion through densely packed molecules [18, 20, 24-26].
Conditions that lead to a EEZ decrease, a deeper solute diffusion through the VELM, can be acheived by treatment with oligosaccharide hydrolyzing enzymes, hyaluronidase [20, 24, 25] and heparinase [27], and by treatment with inflammatory stimuli such as oxidized low-density lipoproteins [28], TNF-α [26] and oxidative stress [20]. EEZ decrease is the early step in the inflammatory cascade associated with I/R injury that results in endothelial dysfunction [10, 12, 13, 17, 24]. Thus, diverse stimuli can alter the VELM structure as a result of a still undescribed change in chemical composition. We hypothesized that ischemia and I/R-induced structural and functional VELM alterations result from biochemical changes that firstly need to be described, later identify the mechanisms behind. This strategy will lead to successful medical treatments.
To isolate with high purity coronary VELM proteins we adopted a novel strategy; in situ VELM protein biotinylation [29]. Experiments were performed in retrogradely perfused isolated rat hearts. Selectively Biotin-labeled coronary VELM proteins from myocardial tissue homogenates were purified and identified. The biotin-labeled coronary VELM proteins were identified either by two-dimensional gel electrophoresis followed by Western blot or by passing the tissue membrane fraction through a Streptavidin-Biotin affinity chromatography column. We found that ischemia and I/R stimuli produced alterations of most coronary VELM protein molecular weight (MW) and their isoelectric point profiles, but I/R insult caused the more drastic changes.
2. MATERIALS AND METHODS (FOR DETAILS SEE SUPPLEMENTAL MATERIAL)
Hearts were removed and coronaries retrogradely perfused [22, 23]. Thereafter, VELM proteins were selectively labeled by intracoronary perfusion of biotin during control conditions. After biotin labeling period, hearts were perfused with buffer and processed for immunohistochemical identification of biotin in tissue sections (brown color).
In three groups of hearts coronary VELM was biotin labeled during control period, thereafter biotin exccess was wash off with PBS. One group of hearts was inmediately processed for tissue protein extraction (control group), a secong group was subjected to 25 min of ischemia and tissue protein extracted (ischemia group) and the third group was subjected to 25 min ischemia, followed by a 30 min reperfusion period (I/R group), thereafter processed for tissue protein extraction. Tissues were homogenized and whole tissue cardiac membranes (CM) were isolated, divided into 2 aliquots to identify coronary VELM proteins, one aliquot was processed for SDS-2D gel electrophoresis and Western blot [30] to identify biotin-labeled VELM proteins (procedure No 1). The second CM aliquot was applied to monomeric avidin affinity column and bound biotinylated proteins were eluted with D-biotin in PBS; VELM fraction (procedure No 2). The SDS-2D gel results obtained with both procedures were the same.
On control coronary VELM protein fraction dot Immuno-blots were performed, dots were incubated with a primary antibody, follow by a secondary antibody coupled to HRP rinsed and DAB was used to develop HRP color reaction. Twelve specific antigens were identified.
3. RESULTS
3.1. Biotinylation of Coronary Vascular Endothelial Membrane Proteins
The heart tissue sections were examined by inmunohistochemical microscopy to verify if biotin was confined to the vascular endothelium. The perfused heart was well preserved morphologically (Fig. 1A, control) and when revealed with DAB intense staining was restricted to the endothelium of all blood vessels, with prominent capillary labeling (Fig 1B and 1C). This indicates the vascular bed was well perfused and most of the vascular endothelial membrane was biotinylated.
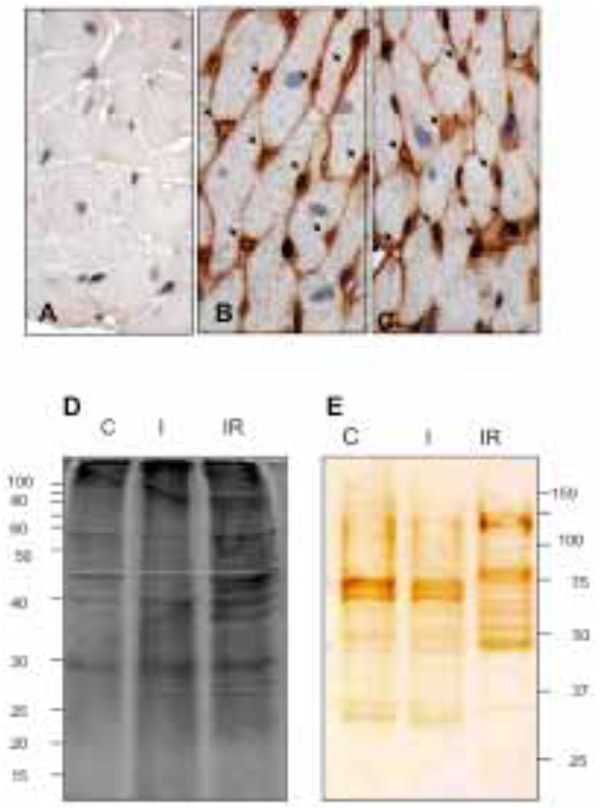
Histological tissue sections: A) non-biotin labeled heart; control. B) and C) biotin perfused heart. Biotinylation; brown color, is restricted to the coronary vascular endothelium. All capillaries were labeled by the biotin (arrows). D) Electrophoretic patterns of CM protein fraction during control (C), I and I/R. E) Electrophoretic patterns of biotinylated coronary VELM. CM protein fractions from SDS-PAGE were transferred to nitrocellulose membranes and coronary VELM proteins revealed by DAB. I/R patterns (E) display more bands of low molecular weight proteins compared to C.
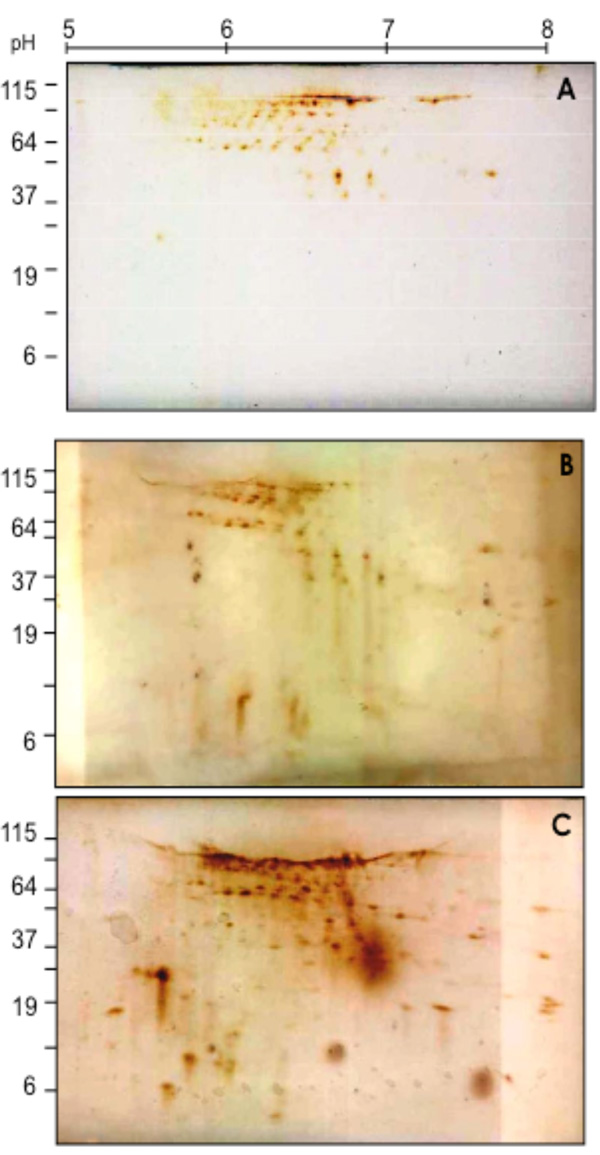
2D-SDS Western blotting patterns of biotinylated coronary VELM proteins during control (A), Ischemia (B) and I/R (C). VELM proteins were biotin labeled during control conditions. Notice that from A to B to C there is an increase in the number of low molecular weight spots.
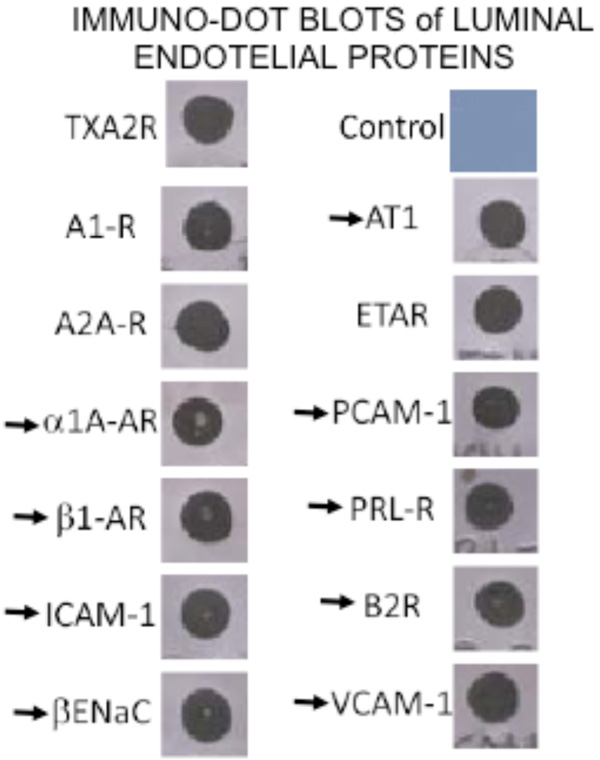
By immuno-dot blot ten G-coupled hormone receptors and three adhesins proteins were identified in the coronary VELM protein fraction. The arrows indicate proteins whose MW-pHi coordinates are known and in the plot (MW-pHi) in figure 4A were identified.
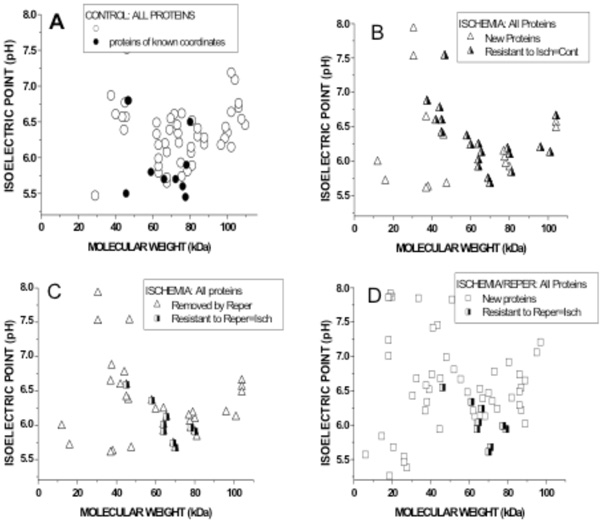
Graphical representation of Molecular weights (abscissae) and Isoelectric pH (ordinates) of biotin-labled VELM proteins. Control coroanry VELM proteins (A), ischemia VELM proteins (B and C) and ischemia/reperfusion VELM proteins (D). Comparison between A and B: (A) all points (circles and half filled triangles, 58 protein-points) show the proteins identified in control. Dots are proteins whose coordinates (MW, pHi) are known and correspond to: α1, β1, AT1, PRL, B2, βEnaC, PECAM-1, VCAM-1, and ICAM-1. (B). All points (triangles and half filled triangles) show the proteins identified in ischemia. Half filled triangles are 20 protein-points that were not affected by the transition from control to ischemia. None of the dots in A appear in B. Comparison between C (=B) and D: (C) all points (Triangles and half filled squares show all the proteins identified in ischemia. Triangle points are proteins that do not appear during ischemia/reperfusion in D. (D) all points (squares and halffilled squares) show the proteins identified in ischemia/reperfusion. Half filled squares are 9 protein-points that were not affected by the transition from ischemia to ischemia/reperfusion. Of these 9 protein-points, 7 also appear in control A, i. e. are proteins resistant to ischemia and I/R.
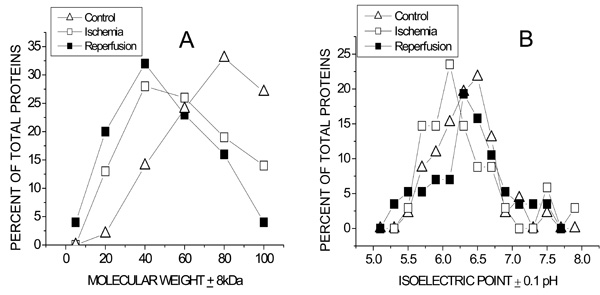
Population distributions of Molecular weight (A) and isoelectric pH (B) of coronary VELM proteins during control, ischemia and ischemia/reperfusion. (A).The greatest number of proteins for control was 80kD, for ischemia and ischemia/reperfusion the peaks were at 40kD. The control curve for MW > 60kD lies above that for ischemia and even lower is the ischemia/reperfusion curve. In contrast, the control curve for MW < 60kD lies below that for ischemia and even lower than the ischemia/reperfusion curve. (B). The control, ischemia and ischemia/reperfusion pHi curves also differ. The greatest number of proteins for control have a pI = 6.40, for ischemia and ischemia/reperfusion the corresponding peaks were at pI = 6.0 and pI = 6.2 respectively.
3.2. SDS-PAGE Protein Profile Ladders
Two equal aliquots of CM proteins were loaded on two separate SDS-PAGE gels and run. Thereafter, one gel was stained with Sypro Ruby (CM, Fig. 1D) and the other gel was blotted to nitrocellulose membrane and developed for biotin (VELM, Fig. 1E). Fig. (1D) show different protein profiles, the CM proteins of control (C), and the two injury conditions, ischemia (I) and I/R heart (IR). The CM protein ladder profiles from control and ischemic hearts show differences that became more striking in the I/R fraction, with increased number of low molecular weight bands. The biotin-labeled VELM protein profile ladders are shown in Fig. (1E). As in the case of Fig. (1D), protein ladder profiles of I/R hearts show striking differences in the number of low molecular weight bands compared to control.
3.3. CM Western Blot Biotin Analysis
To determine in coronary VELM proteins the individual changes induced by ischemia and I/R, the CM proteins subjected to 2D SDS-PAGE were electro-transferred to nitrocellulose membranes and developed with HRP-streptavidin-DAB (Fig. 2). Individual protein profile distributions of coronary VELM differences between individual protein between control (Fig. 2A) and ischemia (Fig. 2B) are noticeable, but more striking differences appear in I/R group (Fig. 2C). These data show that ischemia and I/R injuries affect MW and pHi of many individual coronary VELM proteins of the rat heart.
By dot blot, the following 12 proteins were identified in the VELM protein fraction: the hormone receptors for adenosine A1 and A2A, thromboxane A2 (TXA2), endothelin ETA, adrenergic α1 and β1, Angiotensin AT1, prolactin PRL, and bradykinin B2, the channel βEnaC and the adhesive proteins PECAM-1, VCAM-1 and ICAM-1. All these VELM proteins gave a positive dot blot reaction (Fig. 3). Of this list for α1, β1, AT1, PRL, B2, βEnaC, PECAM-1, VCAM-1, and ICAM-1 their corresponding molecular weigths (MW) and isoelectric points (pHi) have been reported. Protein points with these coordinates were identified in the control biotin-2D-SDS-PAGE results (Fig. 2A).
3.4. Comparison of Protein Profile Distributions from Western Blot Biotin Analysis Studies
To compare quantitatively and directly the differences in coronary VELM bi-dimensional MW-pHi distributions between control, ischemia and ischemia-reperfusion, the coordinates (MW, pHi) of each protein-point was determined and the values plotted as shown in Fig. (4). Keep in mind that all proteins identified via biotin, were labeled during control not during ischemia nor I/R.
3.4.1. Comparison between Control and Ischemia Graphs
The 2 upper figures correspond to control (Fig. 4A) and ischemia (Fig. 4B). In the control graph (Fig. 4A) all points (circles and dots) are 58 protein-points present during control condition. The dots are coordinates reported for the 9 identified proteins (Fig. 4A) mentioned above. In ischemia graph (Fig. 4B) all protein-points (triangles and half filled triangles) present during ischemia are shown. Triangles are protein-points with different coordinates that are not present during control; i. e control proteins whose MW and pHi were chanced by ischemia. But, half filled triangles (20 protein-points) are protein-points that have the same coordinates as protein-points in the control, likely ischemia resistant proteins. These results show that only 20/58 protein-points (34 %) remain unchanged, while 66 % of the protein-points their MW and pHi were altered by ischemia. None of the dots in (Fig. 4A) appear in (Fig. 4B) indicating the coordinates (MW and pHi) of α1, β1, AT1, PRL, B2, βEnaC, PECAM-1, VCAM-1, and ICAM-1 were changed by ischemia. Clearly, transition from control to ischemia results in alteration of the MW and pHi of 66 % of protein-points (ischemia susceptible proteins), while a smaller fraction of protein-points their coordinates remain unchanged (half filled triangles), likely ischemia resistant proteins.
3.4.2. Comparison between Ischemia and I/R Graphs
The 2 lower figures correspond to ischemia (Fig. 4C=4B) and I/R (Fig. 4D). In the left graph (Fig. 4C) all protein-points (triangles and half-filled squares) were present during ischemia. In the right graph (Fig. 4D) all proteins-points (squares and half filled squares) were present during I/R. Transition from ischemia to I/R shows that triangles protein-points (Fig. 4C) are not present in I/R; I/R susceptible proteins, while half-filled squares (Figs. 4C and 4D) are 9 protein-points whose MW and pHi were not chanced by I/R; I/R resistant protein-points. These results show that 9/38 protein-points (23 %) coordinates during ischemia remain unchanged, while 76% of the protein-points their MW and pHi were altered by I/R. I/R cause the appearance of a larger number of biotin-labeld low MW proteins with new coordinates (squares). Here again, transition from ischemia to I/R results in alteration of the coordinates of 76% of protein-points (I/R susceptible proteins), while small number of protein-points their coordinates remain unchanged; likely IR resistant proteins. Of the 9 protein-points; I/R resistant proteins, 7 are protein-points found during control conditions. This implies that only 7/58 protein-points (12 %) in control are ischemia and I/R resistant proteins and the rest 88% their MW and pHi are altered, indicating that likely non-specific hydrolizing mechanisms are activated by ischemia and I/R.
3.4.3. MW and pHi Populations Distribution Plots of Coronary Protein Points During Control, Ischemia and I/R
Since each of the transitions from control to ischemia and from ischemia to I/R are associated with obvious MW-pHi coordinate changes, we decided to analyze the data via MW (Fig. 5A) and pHi (Fig. 5B) population distribution plots. The left graph shows that during control conditions (triangles) coronary VELM proteins have a MW range between 100kDa to 20kDa with the largest proportion of proteins having a MW of about 80kDa and only 13% with a MW less than 60kDa. Ischemic VELM proteins (squares) also have a MW range between 100kDa to 20kDa with the largest proportion of proteins having a MW of about 40kDa and 43% with a MW less than 60 kDa i. e. there is a shift in the distribution curve to the left with fewer number of high MW proteins and more low MW proteins. The changes induced by ischemia are further amplified by I/R. Under this condition (filled squares) the MW range is also between 100kDa to 20kDa with the largest number of proteins having a MW of about 40kDa and there is a further shift of the distribution curve to the left with 51% of proteins having a MW less than 60kDa.
With respect to the pHi protein population distribution plots (Fig. 4B), the majority of proteins from control hearts possess pHi in the 6-6.5 range which shifts to more acidic pHi following ischemia. Reperfusion leads to a shift in pHi distribution towards control conditions.
4. DISCUSSION
Our results show that coronary VELM proteins, “the luminal endothelial glycocalyx” proteins biotin-labeled during control conditions show drastic chemical changes during the successive stages of ischemia and I/R.
Our results of MW population distribution analyses show that a shift toward lower MW proteins with ischemia was further accentuated by I/R. These 2 insults also affected the pHi population distribution (control peak pHi = 6.5), ischemia displaced the group toward a more acidic pH (peak pHi = 6.0) and I/R shifted to pI = 6.3 (Fig. 5B).
In the present work, the biotin coronary VELM protein labeling method by Zhou et al., [29] was modified to be certain that during infusion, the biotin-reactive agent (S-NHS-LC-biotin) was confined to the coronary intravascular space so that only VELM proteins were labeled (Fig. 1). This was achieved by adjusting 2 parameters, the intravascular concentration of the labeling agent was diluted 16 times (from 2 mg/ml to 0.125 mg/ml) to reduce its concentration gradient and the period of S-NHS-LC-biotin perfusion was shortened from 120 to 5 sec to reduce 24 times the diffusion period, followed by a 55 sec wash period (6 cycles). In our hands using the conditions of Zhow et al., [33] resulted in intense labeling of all cell membranes of coronary vascular; endothelial and smooth muscle cells and the cardiac cells membranes (not shown). It is possible that these differences with Zhow et al., work may reside in differences between the vasculature of the 2 tissues. Our methodology detects only those proteins that were labeled during control conditions. Any biotin-labeled proteins identified during ischemia or I/R are to proteins labeled during control except that their MW and pHi were changed by those insults. Our procedures provide information about protein biochemical history from control to ischemia and from ischemia to I/R.
4.1. Possible Activating Mechanisms of the Alteration of Coronary VELM Protein Profiles; An Open Question
MW and pHi alterations of VELM population distributions induced by ischemia and I/R imply that control proteins with each successive transition: control to ischemia (66%) and to I/R (88%), suffer partial hydrolysis either of their proteic (both MW and pHi changes) or/and their oligosaccharide moieties (only MW changes). This widespread hydrolysis include a list of G-protein couple receptors and adhesive proteins (Fig. 3), indicates that hormonal activation of these VELM G-coupled receptors and adhesive proteins is altered. Evidence supports the activation by ischemia and I/R of both proteic and glycan hydrolyzing mechanisms [10, 20, 26, 31-36]. But, the mechanisms of the activation by these insults are only suspect.
Endothelial proteic shedding has been linked to metalloproteinase (MMP) activation and their activation by schemia and I/R in heart is well documented [10, 31-36]. Specifically, MMP activity has been shown to increase following reperfusion and may be responsible for glycocalyx shedding and cleveage of the β2-adrenergic receptor [32, 33]. But, the mechanism of MMP activation is not clear.
In contrast to protease activation by ischemia and I/R, activation of glycosaminoglyclans-hydrolyzing mechanisms have been inferred. Luminal endothelial glycocalyx thickness/EEZ thickness” is reduced in size if heparinase and hyaluronidase [20, 25, 27] are infused. In addition, if hyaluronidate (which competes for hydrolysis with the endogenous hyaluronidate) is infused prior to I/R, the EEZ thickness is not affected [20]. The activation of other types of glycanases by ischemia and I/R is suggested when exogenous lectins upon binding to protein oligosaccharides moieties protect these sites (prevent EEZ reduction) from hydrolysis by I/R activated glycanases [24]. We have recently shown that the level of total glycosaminoglycans of coronary VELM is decreased by ischemia and I/R [36], but the glycanases involved were not identified. Evidence indicates that TNF-α, cytokines and reactive oxygen species play an important role in I/R injury [1, 10, 20, 26, 31-35, 37]. They may be the activating agents of protein and oligosaccharide hydrolyzing enzymes in control VELM seem to be dormant.
4.2. Possible Functional Implications of the Changes of Coronary VELM Properties Induced by Ischemia and I/R
In proteins of the coronary VELM reside multiple functions; solute permeability, hormone reception, flow sensing, inflammation, atherosclerosis, coagulation etc, [7, 12, 13, 17-19, 21-23]. These functions likely are altered from control to ischemia and to I/R because coronary VELM protein MW and pHi profiles between control, ischemia and I/R are different.
Solute endothelial permeabilty is determined by the chemical properties and thickness of EEZ. EEZ thickness suffers important reduction in size during I/R and the associated change in chemical composition (Figs. 3, 4) imply there must be important selective permeability changes [20, 26].
The coronary VELM is an important site of hormone reception [6, 23, 33] and flow sensing [22] a list of G-protein coupled receptors and βEnaC a flow sensor, reside in this structure as shown above. Here we show that these estructures are cleave because their MW-pHi coordinates are altered by ischemia and by I/R, implying that the pharmacology and flow sensing function of VELM are also altered. VELM proteins like PECAM-1, V-CAM-1 and I-CAM-1 associated with inflammatory and coagulation processes [17, 24] their chemistry (MW and pHi) is also altered by ischemia and I/R so must be their function
Much remains unclear about the mechanisms involved in I/R injury. Furthermore, our data are consistent with the idea that a large modification of the glycocalyx chemistry and structure are important steps in the pathological cascade of events leading to I/R injury and infarct size. This indicates that modification of the glycocalyx is a primary component of early I/R effects and should be a therapeutic target in the prevention and/or treatment of I/R injury [1].
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publishers Web site along with the published article.
ACKNOWLEDGEMENTS
Funding by: CONACyT SEP-42567, CONACyT-SALUD 2004-C01-156 and Fulbright-Robles Award #9583.
CONFLICT OF INTEREST
None.

