All published articles of this journal are available on ScienceDirect.

Myocardial Ischemia in Wegener’s Granulomatosis: Coronary Atherosclerosis Versus Vasculitis
Abstract
Wegener’s granulomatosis (WG) is one of the most common small- and medium-sized necrotizing vasculitides that mainly affects the upper and lower respiratory tract and the kidneys. Cardiac manifestations in WG are relatively rare, and their role and place among different causes of mortality remain largely unknown. Substantially increased number of reports describing involvement of all structures of the heart, which underlie conduction disturbances, valvular disease, ischemic heart disease and other potentially serious conditions, underscores importance of comprehensive cardiovascular investigations and monitoring of patients with WG. The majority of previous reports and our current observation distinguish coronary vasculitis and thrombosis as a cause of myocardial ischemia and cardiovascular co-morbidities in WG. It seems plausible that inflammatory processes in this disease, like in some other systemic vasculitidies, do not predispose to accelerated atherogenesis. However, characteristic small- and medium-sized vasculitis still can manifest as myocardial ischemia and infarction. We overview diverse cardiac manifestations and present our own rare case of angina in the oligosymptomatic debut of WG. Importantly, in this case, coronarography failed to reveal atherosclerotic disease or thrombotic occlusion. However, magnetic resonance imaging (MRI) with adenosine test revealed subendocardial ischemia. As a result of immunosuppressive therapy with a steroid and cyclophosphamide, myocardial ischemia disappeared.
INTRODUCTION
In recent years, evidence has accumulated to suggest the crucial role of systemic inflammation and immune dysregulation in enhanced cardiovascular morbidity and mortality in a variety of chronic inflammatory disorders [1-7]. Overexpression of common inflammatory, thrombotic and immune markers alongside with active contribution of disease-specific factors form pathophysiological basis for diverse cardiovascular manifestations: pericarditis, myocarditis, cardiomyopathy, endocarditis, valvular disease, coronaritis, aneurysms, thrombosis and atherosclerotic cardiovascular disease, pulmonary and systemic hypertension, rhythm and conduction disturbances, diastolic and systolic heart failure [1, 8-10]. Moreover, it has been well established that additive effect of classical cardiovascular risk factors, such as smoking, hypertension, dyslipidemia, diabetes, physical inactivity, may substantially increase the risk of vascular events in inflammatory disorders, particularly in systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA) [3, 6, 11-13]. Another important factor increasing cardiovascular risk is a prolonged severe course of an underlying inflammatory condition and associated long-term therapy with high dose steroids, which mediate negative vascular effects through steroid-induced diabetes, hypertension, atherogenic lipid disturbances and obesity [14-19].
Predisposing factors, morphological changes and clinical manifestations of inflammation-induced cardiovascular morbidity and mortality are relatively well investigated in SLE and RA. Though cardiovascular comorbidities in SLE and RA are not limited to ischemic heart disease (IHD), both inflammatory disorders can be viewed as models of enhanced coronary atherogenesis, based on the results of multiple epidemiological, vascular imaging, clinicopathological and pathomorphological studies [20-25]. Importance of these results is difficult to overestimate, especially given the cardiovascular perspectives of anti-inflammatory preventive measures in the general population. Remarkable example, in this regard, is a recent attempt to reduce cardiovascular events in a large population of patients with stable IHD and subclinical inflammation with the use of low dose methotrexate therapy, a first-line drug therapy for RA, in the Cardiovascular Inflammation Reduction Trial (CIRT) [26]. Positive results of this trial will provide strong evidence for inflammatory hypothesis of atherogenesis and pave way for the use of other safe anti-inflammatory drugs in low-grade inflammatory disease states associated with increased vascular risk.
To better understand implications of inflammation in atherogenesis, apart from SLE and RA, cardiovascular manifestations of seemingly less atherogenic disorders, such as primary small- and medium-sized systemic vasculitides (Wegener’s granulomatosis [WG], polyarteritis nodosa, Kawasaki disease, Churg-Strauss syndrome, and microscopic polyangiitis), should be further investigated. Though these disorders are associated with overexpression of multiple inflammatory markers, which cause endothelial damage, there is no direct evidence to categorize them as pro-atherogenic states [27-30]. Differences in atherogenic profiles in SLE, RA and primary vasculitides may be linked to the intensity of systemic inflammation, preferential involvement of disease specific markers, differing in atherogenic properties, and genetic markers with different vasculopathic targets [31, 32]. To further elucidate mechanisms of vasculopathy in systemic vasculitides comprehensive investigation of clinical manifestations of cardiovascular involvement and multinational database of case reports on different types of cardiovascular pathology in each systemic vasculitis are urgently needed. We hereby overview cardiovascular manifestations in WG and analyze own case report.
CARDIAC MANIFESTATIONS IN WEGENER’S GRANULOMATOSIS
WG is one of the most common necrotizing vasculitides. The prevalence of the disease is 3 in 100,000 population with male to female ratio of 3:2, and peak incidence at the age of 50-60 [33]. Etiology still remains unknown. Granulomatous lesions of the upper and lower airways and the kidneys are predominant features of the disease [34,35]. Classical clinical presentation of WG includes ear, nose, throat (ENT) involvement, saddle-nose deformity due to the destruction of the nasal septum, epistaxis, nasal crusting and sinusitis, pulmonary involvement with hemoptysis (alveolar hemorrhage and capillaritis), cough and dyspnea, arthralgia, optic neuritis, peripheral neuropathy, and necrotizing glomerulonephritis [33, 35].
It has long been known that kidney and lung affections are major predictors of mortality, and cardiac involvement is relatively infrequent in WG [35]. In one cohort of 158 patients with WG, cardiac manifestations (predominantly pericarditis) were present in 10 patients (6%), and, of these, 3 patients had coronary artery involvement [36, 37]. The results of autopsy, however, demonstrated that cardiac abnormalities can be present in one third of patients with WG who have no clinical signs of cardiac pathology and die predominantly from non-cardiac causes [33].
In a multiple series of recent observations, clinical significance and potentially lethal consequences of diverse cardiac affections have been recognized. In fact, numerous reports on WG with conduction disturbances, such as complete heart block due to inflammation of atrioventricular node, and arrhythmias with compromised hemodynamics have been published over the past decade [38-43]. Pericarditis, coronaritis, focal myocarditis, cardiomyopathy, noninfectious endocarditis, valvular disease with vegetations, mitral and aortal insufficiency, and systolic heart failure are other frequently reported cardiac manifestations in WG [1, 44-48].
It should be noted that histopathological changes underlying systemic affections in WG occur in the absence of immune deposits, and include neutrophilic vasculitis, necrosis, microabscesses and granuloma [35]. These changes are driven by highly specific for WG antineutrophil cytoplasmic antibodies (ANCA) directed against cytoplasmic compound, proteinase-3 (cANCA). Anti-proteinase-3 antibodies cause degranulation of neutrophils with subsequent release of proteolytic enzymes, damaging the endothelium and triggering vasculopathic cascade [8, 49]. Though it is well-known that endothelial dysfunction due to small vessel disease is characteristic for WG [50], recently published reports, mainly based on the newest imaging techniques, point to the possibility of systemic involvement of larger size vessels, resulting in arterial aneurysms and venous thrombosis [51-57].
In the most cases of WG, atherosclerotic vascular disease, particularly that seen in SLE and RA, has not been documented, reflecting the fact that vascular inflammation and thrombosis, including coronaritis and coronary thromboembolism, as more characteristic for WG [1, 58-60]. In contrary, in a small case-control study, an increase of intima-media thickness (IMT) of the carotid artery was found in patients with WG, interpreted as a sign of accelerated atherosclerosis [61]. The results of this observation, however, need further confirmation in a long-term studies with better defined markers of atherosclerosis (e.g. carotid plaques, angiographically and pathomorphologically confirmed atherosclerotic lesions), since an increase of IMT in a chronic inflammatory disorder can be merely a reflection of inflammation in the vessel wall, but not atherosclerosis [62].
Whatever the underlying mechanism of coronary pathology, in a large cohort of patients with WG, a significantly increased risk of IHD was recently reported (observed to expected ratio of IHD events 1.9, 95% confidence interval [95%CI] 1.4-2.4) [63]. An increased risk of IHD was largely due to the relatively high incidence of myocardial infarction (MI), particularly in those with advanced age at the time of diagnosis, in males, and in those treated with high doses of cyclophosphamide. This study confirmed the results of previous reports, indicating that patients with WG frequently experience ‘silent’ IHD [64,65], which may further contribute to an increase of mortality rates.
Multiple series of case reports and cohort studies analyzed above highlight importance of comprehensive cardiovascular assessment of patients with WG even in clinically asymptomatic cases. It should be also kept in mind that cardiovascular manifestations (e.g. conduction block, MI, heart failure) can evolve in the absence of classical complex of symptoms or in so called ‘limited WG’. We hereby present a rare case of angina in the debut of WG, in the absence of classical cardiovascular risk factors.
CASE PRESENTATION
A 64-year-old woman was admitted to the hospital with angina (CCS II) and signs of heart failure (NYHA III) of two weeks duration. She was examined four weeks prior to the admission. At that time only pain on palpation and symmetrical swelling of her hands were noticed. Diclofenac was administered. The patient had a flu-like illness three months ago, which followed by fatigue, malaise, arthralgia, myalgia, orthostatic dizziness, and morning stiffness. The symptoms improved in response to oral diclofenac.
On physical examination, there was tenderness of wrists and proximal interphalangeal joints. The lungs were clear. A 2/6 grade systolic murmur was audible over the all auscultation points. There were no hepatosplenomegaly, lymphadenopathy.
Importantly, on ECG, there were ST-T changes in I, aVL and V3-6 (Fig. 1). Except for localized pericardial thickening, echocardiography did not reveal any other pathological changes. Abdominal ultrasound and computed tomography (CT) did not reveal pathological changes.
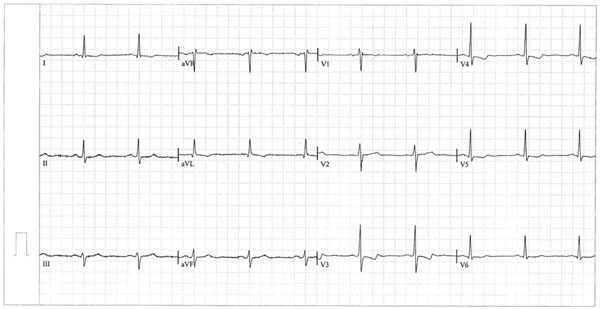
ECG on admission. Regular sinus rhythm, heart axis deviation to the left, ST-T changes of ischemic origin in I, aVL and V3-6.
Blood tests: erythrocyte sedimentation rate (ESR) 90 mm/hour, C reactive protein (CRP) 16.5 mg/L (normal range <9 mg/L), hemoglobin 10.8 g/dL, white blood cells 7,450 per microliter, platelets 434,000 per microliter. Liver and kidney function tests were within normal limits. Urinalysis was without pathological findings.
Test for HIV was negative. Multiple series of blood and urine culture tests were negative.
Antistreptolysin O titer was 1:400 (normal range <1:300), rheumatoid factor - positive, antinuclear antibodies and the Coomb’s test - negative. An enzyme-linked immunosorbent assay was positive for cANCA (43; normal range <20).
On the 3rd day, the patient presented with fever, shortness of breath, paresthesia in both hands and feet. On neurological examination, symmetrical peripheral neuropathy was noted. Chest radiography showed infiltrate in the right lower lobe.
On the 6th day, dyspnea became severe. Arterial partial pressure of O2 (PO2) was 50 mmHg, carbon dioxide (PCO2) 32 mmHg, bicarbonate level 21 mmol/L, O2 saturation 86 %, pH, 7.42. On chest X-ray, pulmonary infiltrates disseminated in both lungs. Bronchoscopy with bronchoalveolar lavage fluid examination revealed multiple erythrocytes; tests for Pneumocystis carinii and Mycobacteria were negative. Lung biopsy revealed alveolar hemorrhage.
On the 7th day, erythematous rash erupted, and skin biopsy performed from the affected area revealed leukocytoclastic vasculitis.
CT of paranasal sinuses was performed, which revealed thickening of the mucous membrane of the left frontal and right maxillary sinuses. Biopsy from the inferior concha was suggestive of nonspecific inflammation.
A bone marrow biopsy revealed mild eosinophilia and plasmocytosis, and otherwise was unremarkable. An open-lung biopsy revealed histological signs of neutrophilic microabscesses, thrombosis, hemorrhage and granuloma characteristic for WG with palisading histiocytes, CD3+ T cells, CD68+ histiocytes, histiocytic giant cells, eosinophils and CD20+ B cells.
The patient was initially treated with combination of cefuroxime, gentamycine and erythromycine. After the diagnosis of WG was made, intravenous methylprednisolone (500 mg daily) and oral cyclophosphamide (300 mg daily) were initiated.
On the 12th day, cardiac MRI was performed (Fig. 2), which showed only mild cardiomegaly with preserved systolic function (left ventricular ejection fraction [EF] 78%) and absence of valvular abnormalities. The aorta and both ventricles were without pathological changes (no signs of old MI). Adenosine infusion, however, revealed subendocardial perfusion defects especially expressed in the septum and posterior wall of the left ventricle.
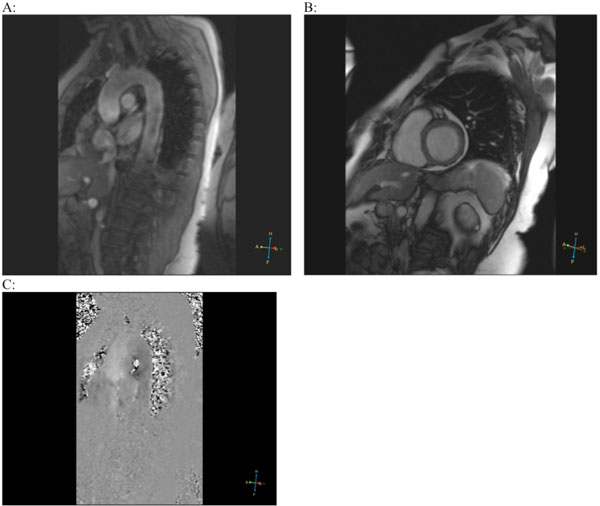
Magnetic resonance imaging (MRI). Ascending and thoracic aorta (A), both ventricles of the heart (B) without pathological changes. After adenosine infusion, endocardial ischemia in the septum and posterior wall of the left ventricle was detected (C).
On ventriculocoronarography, global contractility was not impaired, left ventricular EF was 78%, apex and lateral wall of the left ventricle were unusually rounded; no signs of valvular pathology; except from minor sclerotic changes, coronary arteries were otherwise unchanged (Fig. 3).
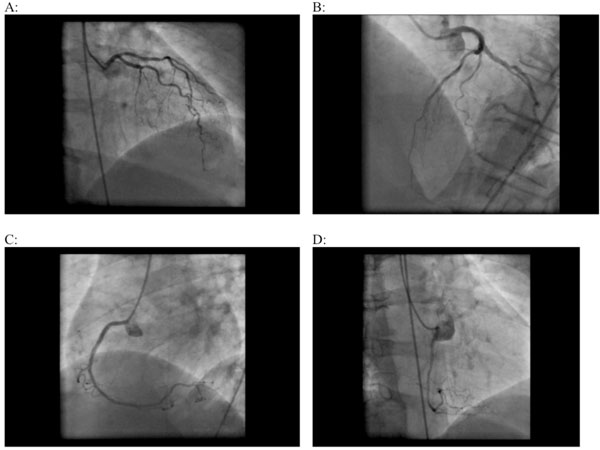
Coronaroangiography. Left (A, B) and right coronary arteries (B, C, D) without visible pathological changes.
The patient was discharged on the 20th day with recommendations on tapering steroid and cyclophosphamide (150 mg daily). Six weeks after the discharge, dyspnea, angina and initially recorded ischemic changes on ECG disappeared, and repeated stress test with adenosine did not show perfusion defects. Renal function tests were within normal limits. Inflammation of paranasal sinuses resolved. Repeated chest CT, performed after 6 months, documented absence of pulmonary infiltrates. Sixteen months after the discharge, vision loss and peripheral neuropathy were residual signs of WG. Cyclophosphamide switched to azathioprine, and prednisone maintenance dose remained at 10 mg daily.
A year later, the patient developed hypertension (>160/100 mmHg), which responded to antihypertensive treatment with angiotensin converting enzyme (ACE) inhibitor and angiotensin II receptor blocker, and the decision was made to start antiplatelet therapy (clopidogrel 75 mg daily) on top of immunosuppressive therapy with prednisone 7.5 mg daily and azathioprine 50 mg two times daily. Repeated cANCA test at that time was still positive (28), ESR was 35 mm/hour, CRP 12 mg/L.
DISCUSSION
Multisystemic affections gradually evolving in this patient were suggestive of WG. The diagnosis, primarily based on the presence of pulmonary infiltrates and biopsy findings, was in accordance with the American College of Rheumatology (ACR) classification criteria of WG [66]. The latter requires presence of at least 2 out of 4 criteria: abnormal urinary sediment, abnormal chest radiograph (nodules, cavities, or infiltrates), oral ulcers or nasal discharge, and characteristic biopsy findings. In our patient, initial diagnostic confusion was due to oligosymptomatic presentation (limited WG) with angina and signs of heart failure. The diagnosis of WG was further supported by positive cANCA test and biopsy suggestive of granulomatous vasculitis. Our case once again underscores the fact that WG can present with myocardial ischemia due to coronary vasculitis [67]. Previously reported cases of coronary artery involvement in WG failed to document clinically significant atherosclerotic disease and proved that myocardial ischemia in WG is mainly due to the occlusion of small- and medium-sized coronary arteries at the cardiac apex [67]. In our case, MRI with adenosine test detected transient subendocardial ischemia in the septum and posterior wall of the left ventricle, which coupled with coronary angiography (no visible changes) and ECG findings (ischemic changes of ST-T), distinguished small vessel disease as a cause of angina. Importantly, angina and ECG changes disappeared in response to the treatment with steroid and cyclophosphamide, which can be viewed as another fact in favor of vasculitic origin of microcoronary abnormalities.

