All published articles of this journal are available on ScienceDirect.

Implications of Genetic Polymorphisms in Inflammation-Induced Atherosclerosis
Abstract
Inflammation is the mainstay of atherosclerosis and is an important governing factor at all stages of the disease process from lesion formation to plaque build-up and final end-stage rupture and thrombosis. An overview of the numerous clinico-epidemiological studies on the association between inflammatory gene polymorphisms and Cardiovascular disease (CVD) and its co-morbidities have shown that the risk associated with any single genotype is modest while the haplotypes, especially those defined on the basis of tag-SNP approach, have better coverage of the gene and show moderately higher impact on disease risk. Nevertheless, even these associations have been inconsistent with low cross-race repeatability. This has been attributed to many plausible causes such as clinical heterogeneity, sample selection criteria, variable genetic landscapes across different ethnic groups, confounding effect of co-morbidities etc. On the other hand, unbiased studies such as the family-based linkage and case-control based associations that have taken into account, thousands of genotypic markers spanning the whole genome, have had the ability to identify novel genetic loci for coronary artery disease. These studies have shown that many inflammatory genes are involved in the regulation of specific biomarkers of inflammation that collectively contribute to the disease-associated risk. In addition, there appears to be considerable cross talk between the different biochemical and metabolic processes. Therefore, consideration of all these factors can build towards an ‘atherosclerotic bionetwork’ that can refine our quest for developing a robust risk stratification tool for cardiovascular disease.
INTRODUCTION
Inflammation plays a pivotal role in the atherosclerotic disease process and is considered to be a primary modulator of disease risk [1]. Inflammatory cells present in the arterial lesions mediate the various processes of plaque formation, progression and vessel thrombosis. Plasma levels of several inflammatory biomarkers are associated with future cardiovascular risk in a variety of clinical settings. Sustained low-grade inflammatory process can be implicated in enhanced atherothrombosis in certain ethnic groups such as the South Asian, Afro-Caribbean and Hispanic and can influence the efficiency of cardiovascular drug treatments (statins as well as aspirin) [2,3]. The seminal role of inflammation in atherosclerosis dates back to more than 70 years and predates the cholesterol hypothesis. The inflammatory perspective gained preeminence in 1999 with the claim that atherosclerosis is primarily an inflammatory disease [1]. Proof of concept for this hypothesis was initially provided by Ridker and colleagues who showed that despite elevated baseline levels of various upstream markers of inflammation and immune response in CAD patients, high sensitive C Reactive Protein (hsCRP) stood out as the best predictor of disease risk. In their study, healthy men who developed a new cardiovascular event during 8-year follow-up showed baseline CRP levels in the top quartile and were three times at higher risk of developing future Myocardial Infarction (MI) and twice at risk for ischemic stroke as compared to men in the bottom quartile [4]. In 2000, the same group showed a similar trend in a 3-year prospective study on post-menopausal women [5]. Over twenty or more large-scale prospective studies have subsequently shown that the relative impact of hsCRP is at least as large as that individually of low-density lipoprotein cholesterol, high-density lipoprotein cholesterol, blood pressure, or smoking. Knowledge of hsCRP was able to correctly reclassify a substantial proportion of "intermediate-risk" individuals into clinically relevant higher- or lower-risk categories in these studies [6]. Risk-prediction models that incorporate hsCRP, such as the Reynolds Risk Score, have improved risk classification and accuracy for global risk prediction, particularly for those deemed at "intermediate risk" by usual algorithms, such as the Framingham Risk Score [7]. Statins, which are primarily advocated as lipid-lowering drug, have been also credited with anti-inflammatory properties [8]. However, clinical trials that are into investigating the long-term benefits of statins such as the JUPITER [9] and PROSPER [10] have shown discrepant findings on the additional value of CRP as a risk predictor in comparison to the prevailing conventional predictors of risk. The outcome of clinical trials is governed by many factors amongst which the design of the trial is a key consideration. Results from other ongoing clinical trials on the efficacy of statin in targeting lipids, inflammation and overall reduction in cardiovascular disease (CVD) risk can bring about better clarity in this matter. A comprehensive overview on the pivotal role of inflammation and its downstream reaction sequale in atherosclerosis from the formation of fatty streak and progression to complex plaque and finally plaque rupture that involves various triggers of the inflammatory process has been lucidly described [11,12].
Interleukin 6 (IL6) is an important pleiotropic cytokine that has a broad range of humoral and cellular immune properties relating to inflammation, host defense and tissue injury [13] and contributes to the clinical evolution of CVD. IL6 is the principle mediator of acute-phase response and primary determinant of CRP production by the liver [14]. Although high levels of IL6 have been implicated with future risk of MI event [5,15,16], presence of high correlation between IL6 and CRP levels undermines the independent value of IL6 in many instances. Additionally, CRP has a inherent stable nature, lacks diurnal variation and is easy to assay and hence scores over many other biomarkers of inflammation as the most valuable predictor of CVD risk [17] and as a useful adjunct for global risk assessment in the primary prevention of CVD [18].
Some of the other common pro inflammatory cytokines include the Tumor necrosis factor 1 (TNFα) produced by the activated macrophages and the Interleukin1 (IL1) released by the macrophages, platelets and injured endothelium. These cytokines act through the recruitment of monocytes / macrophages to the vessel wall thus eliciting increased expression of adhesion molecules such as Monocyte Chemoattractant Protein 1 (MCP-1) and synthesis of prothrombotic factors such as Plasminogen activator inhibitor 1 (PAI1) and von Willebrand factor, that promotes plaque vulnerability and ensuing thrombotic events etc.
The aim of this review is to provide an overview of the possible associations of single nucleotide polymorphisms (SNPs) in inflammatory genes implicated in atherosclerosis. While there are multiple genes that regulate the inflammatory process, we have chosen to elaborate on three key genes in the central inflammatory pathway with emphasis on large studies, primarily to highlight the reported pattern of geno-type-disease association as well as to understand the genetic and environmental factors that influence these outcome such that appropriate study design and methodology may be adopted that adequately address the prevailing lacunae and help in the planning of robust research strategies in future.
INFLAMMATORY GENES POLYMORPHISMS
Single nucleotide polymorphisms in genes relating to inflammation have been linked to enhanced risk of CVD but the risk associated with any one genotype has been considered to be modest. Taking into consideration, the high allele frequency for many of the variants in the constitution of haplotypes, multiple ‘risk alleles’ may have a larger impact on the regulation of gene activity and function [19] as well as in the modulation of clinically significant risk [20]. A number of studies have reported the association of single nucleotide polymorphisms (SNPs) in candidate genes of inflammation with plasma biomarker, CVD and its co-morbidities. An overview on the genotype-phenotype associations of three of the well-studied inflammatory genes that belong to the central inflammatory process are provided in Table 1.
Overview of Genotype-Phenotype Association of some key Inflammatory Genes Variants
| Genes | SNP | rsID | Vascular Events | Surrogate Biomarkers |
|---|---|---|---|---|
| IL6 | -174G>C | rs1800795 | CAD/MI [22, 26, 27] | High plasma IL6 levels [24] |
| CIMT [32] | High CRP levels [22, 52, 53, 55] | |||
| PAD [33] | High Systolic Blood pressure [27] | |||
| Increased arterial stiffness[28] | ||||
| Insulin insensitivity [84] | ||||
| Lipid abnormalities [34] | ||||
| -572G>C | rs1800796 | CAD / MI [38] | High plasma IL6 levels [24] | |
| Plasma IL6, CRP, Fibrinogen [54] | ||||
| CRP | +1444C>T | rs3091244 | CAD [46] | High plasma CRP levels [42, 45, 52, 85, 86] |
| Stroke [87] | Hypertension [47] | |||
| Obesity [50] | ||||
| rs3093058 | High Plasma CRP levels [86] | |||
| rs3091244 | High Plasma CRP levels [49] | |||
| rs1800947 | Arterial pulse-wave velocity [49] | |||
| TNF-α | -308G>A | rs3091256 | CAD [58, 60, 61, 66, 67] | |
| Obesity [63] | ||||
| Insulin resistance [64] | ||||
| Metabolic syndrome [62] | ||||
| Plasma CRP levels [65] |
CAD – Coronary Artery Disease, CIMT – Carotid Intima Media Thickness, CRP- C-reactive protein, IL 6 – Interleukin 6, MI- Myocardial Infarction, PAD – Peripheral Arterial Disease, SNP – Single Nucleotide Polymorphism, TNF- α – Tumor necrosis factor-alpha.
Note: References given in brackets.
IL6 Gene Polymorphisms
Polymorphisms in the promoter region of the IL6 regulate the expression and inter individual variability of plasma IL 6 in a complex, interactive and haplotype-dependent manner [19]. Five common promoter SNPs in the IL6 gene have been shown to be in strong equilibrium with each other using pair-wise Linkage Disequilibrium (LD) analysis [21]. Independently and in combination, these promoter SNPs has been associated with various pathological states of CVD and its co-morbidities. In a recent report on a predisposed cohort of Asian Indians, a risk haplotype, GGAAG, defined by these promoter SNPs was shown to be associated with over 3.7 times risk of CAD (OR 3.676, 95% CI 1.68 – 8.05, P = 0.0017) and a population attributable risk of 21.1% [22].
The –174G>C variant (rsID1800795) has been the most widely studied promoter SNP and both the G and the C alleles have been independently associated with CVD and its co-morbidities. The C allele was associated with higher basal plasma IL6 levels [23,24] and showed over 1.34 times risk of MI in the ECTIM study on 640 patients and 719 controls [25]. Levels of IL6 were shown to reach their peak, 6 hours following bypass surgery, particularly among C allele carriers [24]. Among the WOSCOPS cohort, comprising of 498 MI patients who were followed up for 4.8 year, response to Pravastatin was better among individuals with CC genotype who also exhibited a significant lowered risk of CHD as compared to the placebo treatment group (OR 0.46, 95% CI-0.27-0.79, P=0.019) [26]. The C allele has also been significantly associated with the following disease states - higher systolic BP in men with BMI >24.86 kg/m2; higher risk of CHD among smokers [27]; increased arterial stiffness in the Rotterdam study [28] and elevated HSP60 levels in the KORA 2000 study [29].
On the other hand, the G allele has been associated with increased cerebrovascular events and severe disability post event in the Rotterdam study [30], increased secretion of IL6 and reduced insulin sensitivity in type 2 diabetic subjects, increased correlation between adipose IL6 mRNA expression and insulin resistance in morbidly obese individuals [31], increased Intima-Media Thickness ( IMT) in middle-aged men without exercise induced ischemia [32], risk of peripheral arterial disease (PAD) in Ty2DM patients [33] and abnormalities in lipid metabolism [34].
In contrast to the above findings, the large MONICA survey on Bavarian residents have shown no association of the –174G>C SNP with any of the traditional risk factors nor prevalence of MI [35], while the Turkish case-control report on a small cohort of 120 CAD patients and 105 controls showed that this polymorphisms does not contribute to the risk stratification [36]. In one of the largest report to date on 2691 Ty2DM patients and 2238 controls from the Nurses’ Health Study and the Health professional follow-up study, no association was shown between diabetes and any of the five IL6 promoter SNPs, which was additionally verified by comprehensive meta analysis ([21]. Genotype-phenotype studies on Ischemic stroke patients exhibited a similar variability in the pattern of association of either the G or C allele of the –174G>C [37] with stroke.
With regard to the other promoter variants in the IL6 gene, the G allele of the –572 G>C SNP was associated with MI in a Han Chinese population [38], with a RR of 1.652 for CHD in another Chinese study [39] and with plasma IL6, CRP and fibrinogen in CAD patients in a Korean study [40]. Ex vivo stimulation of leukocytes by LPS showed elevated baseline levels of IL6 among carriers of the G allele in a study involving both –597G>A (rs1800797) and –174G>A SNPs [41].
The underlying reasons for these inconsistencies in study findings are manifold. Primarily, IL6 has a complex physiology with both pro-and anti-inflammatory properties and complex pattern of transcriptional regulation. Presence of clinical heterogeneity, lack of stringent phenotype definition of cases and controls and presence of co-morbidities hinder the identity of true causal verses casual effects. Other plausible issues relating to genetic factors such as Hardy Weinberg Equilibrium (HWE), allele frequencies, influence of neighboring genetic variants and time of sample collection can also influence the outcome of the study findings.
CRP Gene Polymorphisms
Baseline CRP level is influenced by genetic, environmental and clinical factors [42]. CRP shows a heritability of 27%- 40%, indicating considerable influence of genetic regulation on this trait [43,44]. In an in-depth analysis of over thirteen lead SNPs selected using the tag-SNP approach in 3301 participants from the Framingham Heart Study (FHS), Kathiresan S et al. identified one triallelic SNP +1444C>T>A (rsID3091244) in the promoter region that was significantly associated with high CRP levels (p<0.0001). However, this SNP was able to independently account for only 1.4% of the variation in CRP levels while twelve of the clinical correlates explained up to 26% of the variability, BMI being prominent among them [45]. In yet another large investigation of over 7159 subjects from the Third NHANES cohort comprising of mixed ethnic groups, two SNPs, rs3093058 and rs3091244 were associated with high CRP levels and two SNPs, rs1205 and rs2808630 with low CRP levels. After adjusting for covariates and post-hoc analysis, only rs3091244 SNP was associated with prevalent CAD, particularly in the non-Hispanic White population [46]. In a more recent analysis of data pooled from 5 different studies comprising of 4610 CAD patients and over 14,000 non-CAD subjects, it was observed that although the rs3091244 variant was significantly associated with high CRP levels, there was no association with risk of CHD, particularly after adjusting for confounders. This SNP along with other variants have been associated with CRP levels and hypertension in the Turkish study [47], with Ischemic stroke in a Chinese study [48], with arterial pulse-wave velocity in a Japanese cohort [49] and with obesity in a Taiwanese cohort [50]. However, in the Segovia study on a Spanish population, although four SNPs (rs1417938, rs1800947, rs1130864, rs1205) were significantly associated with CRP levels after adjusting for clinical variables and was able to explain up to 2.9% of the total variability in CRP levels, interestingly, this did not include the better known rs3091244 SNP [51]. Regulation of plasma CRP levels has been additionally associated with promoter polymorphisms in the IL6 gene in various studies [52-54]. A genome-wide association study was performed in 6345 healthy women employing 336,108 SNPs, considered as potential determinants of plasma CRP in the Women’s Genome Health Study. This study identified seven novel gene loci, including the HNF-1 (hepatic nuclear factor 1alpha) and GCKR, belonging to various inflammatory and metabolic pathways that showed significant genome wide association with CRP levels [55]. In a comprehensive two-stage analysis of a large combined cohort from the PARC and the CHS study population, one SNP, rs7953249, in the HNF-1 A gene was significantly associated with plasma CRP levels after adjusting for all potential clinical covariates and rigorous statistical analysis [56]. A 17A repeat allele variant in the FLAP gene has been associated with high CRP levels in CAD patients in a Taiwanese study [57]. Thus, the paradoxical findings on the association of CRP gene polymorphisms with CVD paint a similar picture to that of the IL6, wherein complex regulatory mechanisms, confounding interactions with conventional risk factors, variable genetic landscape of the geographic population under study and presence/absence of HWE that have not been adequately addressed in some of these studies, could possibly explain the vagaries in the study findings.
TNF-α Gene Polymorphisms
The tumor necrosis factor is yet another important cytokine that participates in the inflammatory reaction and has been attributed with modulation of CVD risk. Polymorphisms in the TNF-α and its receptor genes (TNFR1&TNFR2) and the gene for TNF-alpha converting enzyme (TACE or ADAM17), that mainly controls the rate of its shedding from cell surface, have been investigated in the various studies. The –308 G>A (rs3091256) promoter variant has received the most attention and has shown contradictory associations in the development of CAD [58-60] in a population specific manner. Others have reported association with CAD in the presence of specific co-morbidities such as obesity, high systolic BP and plasma insulin levels [61] as well as the metabolic syndrome phenotype [62]. The A allele has also been independently associated with obesity in a Spanish study [63], insulin resistance in obese subjects in an Australian study [64] and plasma CRP levels in the HERITAGE study [65]. Variations in the ADAM17 gene have been shown to be marginally associated with high plasma TNF- α levels as well as risk of cardiovascular death in a prospective cohort of 1400 individuals enrolled in the ATHEROGENE study [66]. Analysis of the TNF superfamily 4 (TNFSF4) gene variation showed limited association of these variants with CVD in Women in a prospective, all-white, WHS cohort comprising of 344 incident CVD cases and 2254 control participants who were followed up for10 years [67,68].
With regard to some of the other pro-inflammatory cytokine genes, the Interleukin-1 (IL1) gene cluster and the IL18 and its receptor (IL18R1 & IL18RAP-receptor accessory protein) have been investigated in a few studies. While there is no debate that their levels may be elevated in patients with CAD who exhibit a strong inflammatory response, the direct association of genetic variants within these genes with CVD has been largely negative [69,70] except in limited studies [71]. Baseline levels of IL-18 were higher among healthy women in a nested case-control study who subsequently developed CVD. The RR for future CVD event was 2.53 among individuals in the highest quartile of IL-18 levels as compared to those in the lowest quartile. While that risk was attenuated after adjustment for traditional cardiovascular risk factors, those with IL-18 levels at or above the 90th percentile threshold remained at elevated risk even after adjustment [72]. Similarly, while IL10 has been known to exert an anti-inflammatory effect and lowered risk of coronary events in patients with unstable angina [73,74], no direct association has been reported with the IL0 polymorphisms in the modulation of CAD risk.
Family-based linkage studies and genome-wide association studies have gained immense popularity and attention in the last decade. Such studies involving a consortium of multiple reputed institutions have a combined power of efficient, high-throughput technologies backed by ample financial resources to scan over 100 K – 1 million variants spanning across the entire genome in an unbiased manner, and have been able to identify interesting and novel loci associated with CAD [75-78]. Studies that have involved a restricted clinical phenotype such as MI have had better success than others that have investigated an ‘all-inclusive CAD phenotype’, which may represent gradient as well as overlapping clinical presentations with inadequate distinction between them. Linkage studies on biomarkers of vascular inflammation, namely CRP, IL6, MCP-1, sICAM in the Framingham Heart Study has shown that multiple genes such as SELE, SELP and CRP on chromosome 1 that influence their levels and may have a potential role in the development of CVD [43]. Analysis involving quantitative trait loci (QTL), as in the Quebec family study, has identified novel loci for CRP on 12p11.23 (P = 0.001) and 12q15 (P = 0.0005) and for IL-6 on 14q12 (P = 0.002). However, none of these loci remained significant after adjusting for BMI [79]. In the HERITAGE family study, strong evidence of linkage was observed on chromosome 20q13 and suggestive evidence on several other chromosomal loci implicating multiple loci for regulating inflammatory response [80]. Genome wide scan with microsatellite markers in hypertensive subjects have identified QTL for CRP on chromosome 10q22 and 21 [81]. While ‘stand-alone’ association studies do contribute to the general knowledge-pool, such high-powered, hypothesis-free association studies have the strength in sample size as well as sophisticated statistical methods that can adjust for the effect of multiple confounding factors and can therefore add tremendous value towards delineating of the molecular basis of CVD.
An overview on these findings carried out mainly in Caucasian populations, however reveal that there is limited cross-race repeatability. While clinical heterogeneity, variable pathological settings arising out of confounding co-morbidities and ethnicity have been some of the key issues on one hand, technical matters such as SNP selection based on the Hapmap data that has limited SNP coverage, have added to the differences in study findings.
It is worthwhile to ponder that various implicated putative CAD loci that have attained genome wide significance have been linked to both cholesterol metabolism (PCSK9, LDLR, LPA) [76,78,82] and to inflammation (HNF-1, CXCL12) [75,76]. Clinical studies have shown that while lipid levels relate to plaque build-up in the artery, inflammatory response preempts plaque instability, rupture and thrombosis. However, both lipid metabolism and inflammation are complex, independent yet interdependent processes that can modulate CVD risk in a clinical setting. While the various statin trials on one hand suggests this link, the experimental observations published by Kleeman et al. in 2007 serve as a true litmus test to this ongoing relationship between lipids and inflammation [83]. Through application of systems biology tools of transcriptomics and metabolomics, the authors have shown that based on the level of cholesterol diet intake in the ApoE Leiden transgenic mice, the liver switches from a resilient ‘low cholesterol stress state’ to a predominantly ‘inflammatory state’ and this intricate switching is controlled by common transcriptional regulators, thereby providing a rationale for an interrelationship between the two processes.
CONCLUSIONS
From the various genetic studies discussed above, it appears that consideration towards a study design that adequately addresses issues such as sample size, sample composition based on stringent clinical criteria, allelic frequencies, HWE, LD distribution and mode of inheritance would be critical factors that can govern the successful outcome of the study. In addition, the future course of design for improved risk stratification should bear in mind the presence of a multiple loci /genes that include overlapping atherothrombotic pathways, which act in tandem to modulate the inflammatory milieu of the individual against the background of well-established risk factors (Fig. 1). Sensitive proteomic platforms such as the bead-based arrays facilitate the simultaneous detection of multiple, low abundant proteins in circulation which when combined with the genetic profile generated from ‘chip-based’ gene arrays, can define an ‘atherosclerotic bionetwork’ that can refine our quest towards development of a robust risk stratification tool for cardiovascular disease.
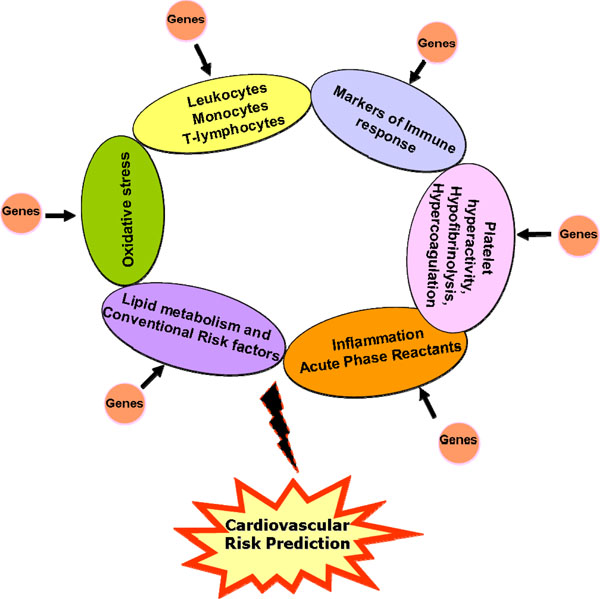
Schematic model incorporating various atherothrombotic risk factors and associated genes for enhanced cardiovascular risk prediction
ABBREVIATIONS
ACKNOWLEDGEMENTS
We thank the trustees of Thrombosis Research Institutes in Bangalore, India and London, UK for their constant support and encouragement. We gratefully acknowledge the financial assistance provided by the Tata Social Welfare Trust and the Department of Biotechnology from India as well as the Weston Foundation and the Emmanuel Kaye Foundation in UK to the Thrombosis Research Institute India and its numerous research studies.

