All published articles of this journal are available on ScienceDirect.

A Case Series of Hypertrophic Cardiomyopathy Conducted in Vietnam Revealing a Novel Pathogenic Variant of the TNNT2 Gene
Authors Info & Affiliations
Abstract
Background:
Hypertrophic Cardiomyopathy (HCM) is one of the leading causes of sudden cardiac death in adults.HCM is inherited in an autosomal dominant manner; however, the genetic etiology of the disease is not fully explained and studies on the hereditary characteristics in family trees are still underway.
Methods:
Ten HCM patients and 31 of their relatives were recruited. Targeted sequencing for 4 HCM related-genes, including MYH7, MYBPC3, TNNT2, and TNNI3, using targeted next-generation sequencing (NGS) was carried out. Demographic, clinical, electrocardiography, and echocardiography characteristics were also characterized.
Results:
Among the 10 HCM patients, 5 were identified with the HCM pathogenic variants in MYH7 (3 patients), MYBPC3 (1 patient), and TNNT2 (1 patient) genes. Eleven out of 31 relatives from these 5 genotype-positive patients carried the same pathogenic variants. We found the novel c.822-2 A>G variant in the splicing site of the TNNT2 gene responsible for HCM disease in a family with 7 subjects genotype positive and 3 others who suffered from sudden cardiac death.
Conclusion:
This case series highlighted the importance of genetic testing for clinically confirmed HCM patients and family members. The genetic information can be used as a molecular marker to complement the clinical presentation in the diagnosis of HCM, as well as a prognostic tool for the patients and their family members.
1. INTRODUCTION
Hypertrophic cardiomyopathy (HCM) is recognized as the most common monogenic cardiovascular disease with a prevalence of 1:500 in young adults. It is also one of the leading causes of sudden cardiac death in young adults [1, 2]. HCM is defined as the thickening of the myocardial wall in any segments of the left ventricle (LV) in the absence of any other causes [3]. HCM is inherited as an autosomal dominant genetic trait that could be transmitted to offspring with a proportion up to 50%. Recent studies have indicated that 60% of HCM patients could be identified with the sarcomere disease-causing variant [4, 5].
The genetic etiology of HCM is not fully explained and studies on the genotype-phenotype correlation are still underway. Up until recently, 11 genes have been identified and extensively studied for their pathogenicity in HCM. Among them, pathogenic variants in a β-myosin heavy chain (MYH7) and cardiac myosin-binding protein C (MYBPC3) genes are the most commonly associated with HCM, followed by Cardiac Troponin T (TNNT2) and Cardiac Troponin I (TNNI3) genes [6, 7].
Recent existing data have been mostly obtained in Caucasian populations while there is a dearth of studies conducted on the Vietnamese population, particularly the population in the North of Vietnam. The study of Thu et al. focused on 23 disease-related genes has demonstrated a prevalence of HCM-related gene variants in Vietnamese people [8]. However, the hereditary characteristics in families as well as the clinical characteristics of probands and their relatives have not yet been clarified. Therefore, we carried out this study on a series of patients and family members, with an extended Next Generation Sequencing panel covering the 4 most prevalent HCM-related genes (MYH7, MYBPC3, TNNT2, and TNNI3). We also discussed the extent of penetrance and phenotype-genotype correlation of the variants found in our study and performed a literature review to assess differences across populations.
2. METHODS
This study was conducted at the Vietnam National Heart Institute (VNHI), Bach Mai hospital, Hanoi, Vietnam, from July 2018 to August 2019. Ten patients diagnosed clinically with HCM were recruited together with 31 family members. The study protocol was approved by the Ethics Committee of Hanoi Medical University (Hanoi Medical University Institutional Review Board, Hanoi, Vietnam, reference number: IRB00003121). The study was abided by the Declaration of Helsinki in regards to the study involving human subjects. Informed consent was obtained from all participants and parents (for patients under 18 years old).
2.1. Clinical Assessment
Patients were clinically diagnosed with HCM by experienced cardiologists and echo-cardiologists at the Vietnam NationalHeart Institute (VNHI). The diagnosis criteria were based on the 2014 (European Society of Cardiology) ESC guidelines defining it as the wall thickness of any left ventricle (LV) myocardial segments ≥15mm with no other causes explaining the hypertrophy. For children, the diagnosis of HCM was based on an LV wall thickness of 2 standard deviations greater than the predicted mean (z-score>2) [9, 10]. Other clinical data, including symptoms, medical history, and family history, were also recorded. A 12-lead resting electrocardiogram was performed on all subjects (Nihon Kohden Model 1250A Cardiofax S, Nihon Kohden, Tokyo, Japan). The presence of atrial fibrillation (AF), LV hypertrophy, left or right bundle-branch block, left atrial, and/or ventricular enlargement, abnormal Q waves, and non-specific ST-T segments were recorded. Patients with suspected paroxysmal arrhythmias (paroxysmal atrial fibrillation (PAF), ventricular tachycardia, premature ventricular contractions (PVC), or atrioventricular block) were confirmed with ambulatory electrocardiogram monitored for 24 h. Echocardiography evaluation for patients and relatives was performed using GE Vivid E9 Ultrasound System (GE Healthcare, Boston, US) and EchoPAC Clinical Workstation Software version V202 (GE Healthcare, Boston, US). All the indexes, including measurements of LV dimensions and wall thickness (septal, posterior, and maximal left ventricular thickness), LV ejection fraction, right ventricular hypertrophy and right ventricular dynamic obstruction, LV diastolic function, left atrial volume index, pulmonary artery systolic pressure, dynamic obstruction at rest and combined with the Valsalva maneuver, mitral valve, and papillary muscle functions were evaluated according to recommendations of the 2011 American Society of Echocardiography [11].
2.2. Genetic Analysis
Upon enrollment, blood samples were obtained from each subject and collected into sterile EDTA (ethylenediamine tetraacetic acid) tubes for genotyping and laboratory analysis. Genomic DNA was isolated from a 2ml peripheral blood sample obtained from each subject using the Wizard® Genomic DNA Purification Kit (Promega, Madison, USA). DNA was stored at -80°C until the time of analysis. Samples were sent for Next-Generation Sequencing (NGS) of a small panel of 4 genes, including MYH7, MYBPC3, TNNT2, and TNNI3. Sanger’s sequencing was the method of choice for verification of identified variants and testing of family members (Primers’ sequence for Sanger’s sequencing is available upon request). PCR amplification product (100-150 ng starting DNA) was obtained for each sample. After agarose gel discrimination, the PCR product was purified with Gel Purification Kit followed by sequencing using Big Dye Terminator V3.1 (Applied Biosystems, Massachusetts, US). Results were analyzed by CLC Main Workbench Software (Qiagen, Hilden, Germany).
2.3. Variant Analysis
Genetics variants were evaluated for their pathogenicity based on the guideline of the American College of Medical Genetics [12]. Appropriate in silico tools and public domain libraries (ClinVar – Clinically relevant Variant, HGMD - Human Gene Mutation Database; LOVD - Leiden Open Variation Database) were utilized to confirm whether variants were previously reported [12]. Pymol was utilized to visualize the affected protein (as well as the location of the pathogenic variants (comparing the native and mutant amino acids) [13].
3. RESULTS
Clinical characteristics, ECG, and echocardiography findings of 16 subjects having genetic positive results are shown in Table 1. We identified four reported pathogenic variants and a new variant of TNNT2 in 5 families. The age of diagnosis for probands with confirmed pathogenic variants was 29 ± 14.7 years for 5 patients with HCM-related pathogenic variants and 64.8 ± 4.5 years for the group without any suspected pathogenic variant; the difference was significant (p=0.02). Other characteristics of the two groups of patients are shown in Table 2.
| S.No. | Mutation |
Relation- ship |
Age | Sex | Symptoms | Cardiac Phenotype | ECG | LVWT (mm) | EF (%) | LVOT, mmHg | Management |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | p.T1377M MYH7 |
Proband | 62 | F | Dyspnea, syncope | Septal hypokinesis (after ASA) | AF, LBBB | 20 | 58 | 60 | BBs and ASA |
| Son | 40 | M | DOE, chest discomfort | Sigmoid septum with no obstruction | NSR, NSST-T | 18 | 73 | 5 | BBs | ||
| Grandson | 11 | M | None | Normal | NSR | 8 | 82 | 9 | None | ||
| 2 | p.R782H MYH7 |
Proband | 39 | M | Dyspnea, oedema, syncope | Septal hypokinesis (after ASA) | BAV III PMR VT in Holter 24h |
22 | 37 | 55 | BBs, ASA, and ICD implantation |
| Brother | 27 | M | DOE, chest discomfort | Septum and apex hypertrophy, no obstruction | NSR, NSST-T | 19 | 61 | 8.5 | BBs | ||
| 3 | c.822-2A>G TNNT2 * |
Proband | 25 | F | DOE, chest pain | Both ventricular hypertrophy with LVOT | RBBB | 24 | 58 | 110 | Medical treatment for heart failure, BBs and ASA |
| Father | 54 | F | DOE, syncope | RV hypertrophy | NSR RVH |
15 | 65 | None | Medical treatment for heart failure | ||
| Aunt | 52 | M | DOE | RV hypertrophy | BAV III | 14 | 68 | None | Pace-maker implantation, BBs | ||
| Aunt | 37 | F | DOE | Septal and RV hypertrophy | NSR NSST-T |
13.4 | 66 | 10 | BBs | ||
| Uncle | 41 | M | None | Normal | NSR, NSST-T | 7 | 80 | None | None | ||
| Cousin | 3 | F | None | Normal | Normal | 5 | 71 | None | None | ||
| Cousin | 1 | M | None | Normal | Normal | 6 | 65 | None | None | ||
| 4 | p.D770N MYBPC3 |
Proband | 18 | F | DOE, palpitations | Sigmoid septum, no obstruction | NSST-T PAF in Holter 24h |
24 | 74 | 12 | BBs, Acenocoumarol |
| Father | 52 | M | None | Normal | NSR RBBB |
10 | 69 | None | BBs | ||
| 5 | p.R1114H MYH7 |
Proband | 30 | M | Dyspnea, chest pain, Syncope | Symmetric hypertrophy | NSTT-T | 31 | 80 | 50 | BBs and ASA |
| Father | 53 | M | DOE | Sigmoid septum, no obstruction | NSR, RVH |
19 | 82 | None | BBs |
* Novel mutation in TNNT2 gene.
| Proband Clinical Characteristic | Confirmed Pathogenic Variant (n=5) | No Genetic Findings (n=5) | p |
|---|---|---|---|
| Gender, male/female | 2 males/3 females | 3 males/2 females | ns |
| Age at diagnosis, years | 29 ± 14.7 | 64.8 ± 4.5 | 0.02 |
| Syncope/Presyncope | 3 | 0 | ns |
| Cardiac symptoms (NYHA ≥ 2/CCS ≥ 2) |
4 | 4 | ns |
| Family history of SCD | 3 | 1 | ns |
| Family history of HCM | 5 | 3 | ns |
| Maximal LVWT, mm | 17.9 ± 10 | 17.1 ± 1.9 | ns |
| Severe hypertrophic >20 mm | 4 | 3 | ns |
| LVOT pressure, mmHg | 18.2 ± 12.5 | 55.6 ± 48 | 0.08 |
| LVOT ≥30 mmHg | 4 | 4 | ns |
| LVOT ≥50 mmHg | 4 | 2 | ns |
| SAM | 4 | 4 | ns |
| Heart failure, LVEF <40% | 1 | 0 | ns |
| ECG abnormalities | 5 | 4 | ns |
| Treatment | |||
| Medications only | 1 | 3 | |
| ASA/Septal myectomy | 4 | 2 | ns |
| ICD | 1 | 0 |
3.1. Results of Genetic Analysis
By fully sequencing 4 common genes related to HCM (MYH7, MYBPC3, TNNT2, and TNNI3), we identified genetic variants in 5 families that can be attributed to HCM. The variants were p.T1377M, p.R783H, p.R1114H on MYH7, p.D770N on MYBCP3, and c.822-2A>G on TNNT2. Reconfirmation using Sanger’s sequencing was performed on the proband as well as the family members to identify that all 5 variants found were in the heterozygous state and exist in at least 2 members of each family. The variants found are outlined in Table 1; the primer for Sanger’s sequencing is available from the authors upon reasonable request. Four out of 5 variants have been identified in patients with HCM (p.T1377M, p.R783H, p.R1114H on MYH7, and p.D770N on MYBCP3) and one newly identified variant c.822-2A>G on TNNT2 was identified. However, we still performed an assessment based on the ACMG (American College of Medical Genetics) guidelines [12] to classify the pathogenicity of variants. We assessed four reported variants based on ACMG criteria matched with previous pathogenicity reports available on databases, such as Clinvar, HGMD, or LOVD. The full criteria and classification are included in the Supplementary Tables. Here, we identified a novel variant not previously reported in patients with HCM, c.822-2A>G on TNNT2.
3.1.1. Synopsis of the Novel TNNT2 Pathogenic Variant
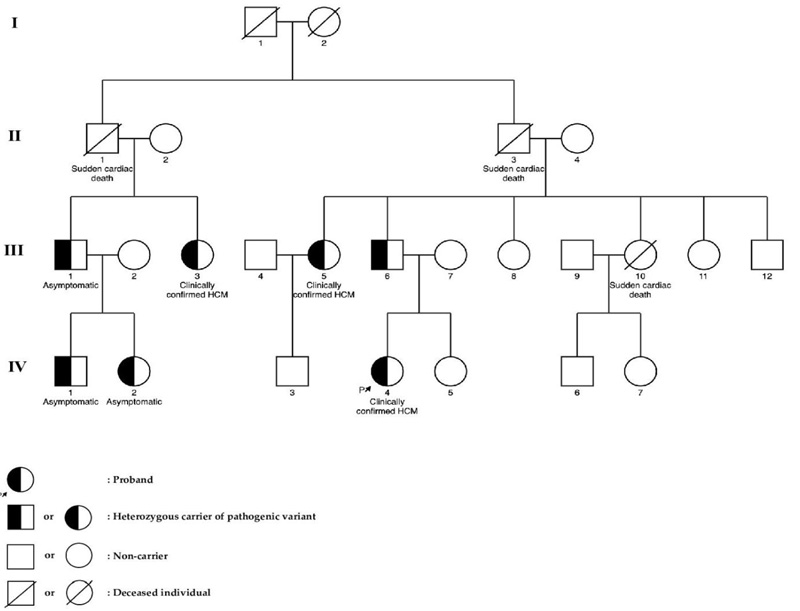
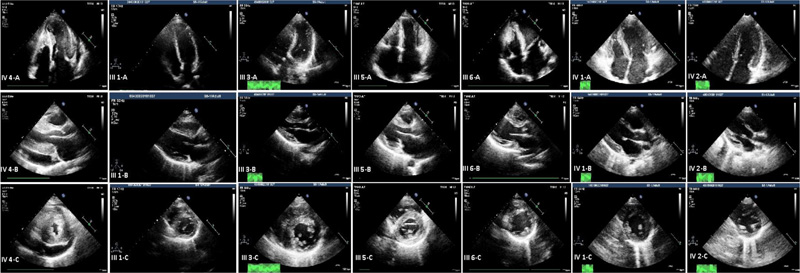
Fig. (1) shows the pedigree of the family with the novel c.822-2A>G variant in the TNNT2 gene. The proband (subject IV.4) was a 25-year-old woman who had dyspnea on exertion and chest pain. Echocardiography showed left ventricular hypertrophy involving the septum, anterior, anterolateral walls, and also the apex with severe LV outflow tract obstruction (pressure gradient, 110 mmHg). She was treated with alcohol septal ablation (ASA) and beta-blockers (BBs), and the symptoms were alleviated (Fig. 2).
At the time of research, 11 of her relatives were screened for HCM. Three of them, including the proband’s father (subject III.6) and 2 of her aunts (subject III.3 and subject III.5), were clinically confirmed to have HCM and also carried the same pathogenic variant as the proband. Meanwhile, her uncle (subject III.1) and his children (subjects IV.1, IV.2) who were also a carrier of this variant had no clear indication of HCM. The patient’s mother (subject III.7), her sister (subject IV.5), two of her aunts (subjects III.8 and III.11), and her uncle (subject III.12) did not have any manifestations of HCM and were genetically negative as confirmed by using the Sanger sequencing method.
Three other relatives (subjects II.1, II.3, and III.10) were reported to have suffered sudden cardiac death (SCD) at the age of 40, 63, and 30 years, respectively. All of them were asymptomatic and did not have a history of any diseases prior to their death. Clinical characteristics of subjects carrying the c.822-2A>G variant in the TNNT2 gene are shown in Table 1.
4. DISCUSSION
In this study, HCM-causative variants of 4 genes, including MYH7, MYBPC3, TNNT2, and TNNI3, which encoded for the beta-myosin heavy chain, myosin-binding protein C, cardiac troponin T, and cardiac troponin I, respectively, were found to be associated with HCM in our cohort. Among 41 individuals from the families of 10 patients, 5 out of 10 patients (50%) carried a single variant related to MYH7, MYBPC3, and TNNT2 genes and 11 (35.5%) of their relatives had the same variants. A novel c.822-2A>G variant of the splicing receptor zone of TNNT2 was found in a family with 7 individuals positive with HCM genetic testing but only 4 out of these 7 individuals had HCM phenotype.
The prevalence of attributable pathogenic variants in our cohort having familial HCM was lower than the reported prevalence in the US and French cohorts, which were 54.2% and 60.6%, respectively [13, 14], but higher than other cohorts in Portugal, Taiwan, Finland, and Japan, which accounted for 28%, 34.2%, 38.2%, and 43.8% [15-18], respectively. Recently, a study conducted on Vietnamese HCM patients in the South of Vietnam found a panel of 23 HCM-related genes which pointed out that 43.4% of patients diagnosed with HCM carried variants mostly focused on MYH7, TPM1, and TNNT2 genes, which were linked with more severe clinical manifestations [8]. However, the difference in prevalence might be due to the limited number of cases in our cohort as well as the difference in gene panel used.
Many previous studies involving patients with familial HCM in Caucasian and Asian populations found mostly variants of MYBPC3 and MYH7. Our study noted that in 2 families which carried pathogenic variants of the MYH7 gene (p.T1377M and p.R782H), sudden cardiac deaths of family members occurred more frequently. Fujita et al. [19] reported Japanese patients with HCM carrying variants in the TNNT2 gene that were associated with a risk of sudden cardiac death in youth. In our cohort, family number 3 were found to carry the novel variant c.822-2A>G in the TNNT2 gene, which might have caused the SCD of 3 family members. In this family, 6 family members were carrying the same c.822-2A>G variant along with the patient, but only 3 of them expressed the HCM phenotype, including the patient’s father and her 2 aunts. Three other individuals were genotype positive – phenotype negative, but 2 of them (subjects IV.1, IV.2 in Fig. (1) were under the age of 5 years.
TNNT2 gene has 16 exons, and the novel c.822-2A>G mutation occurs at the exon-16 splice acceptor site, thus affecting only the ultimate C-terminal part of the protein. We were able to find one study that modeled in vivo human TNNT2 intron 15 mutation that causes two different mutants splice products of TNNT2 [20, 21]. The location of their pathogenic variant was in close proximity to the variant in our study (c.822-2G>A); in their study, the resulting morphant splice product excludes human Exon 16, causing a disruption in the C-terminus of the zebrafish TNNT2 protein at the identical position to that mutated in humans, and is thought to be involved in the interaction with tropomyosin. The morphant ventricles exhibit restrictive physiology, and diminished contractility and defective tnnt2 splicing cause sarcomeric disarray in the embryonic heart, leading to HCM.
The genes we chose to investigate in our panel were more frequently reported to be associated with HCM, and each has a direct role in the heart muscle constitution and function. However, we suspect that there would be other potentially novel pathogenic genes or pathogenic variants in the non-coding region which could be identified with an expanded panel of Whole Exome or Whole Genome Sequencing. In this study, due to the number of cases and short observation time, we were unable to gauge the difference in disease severity of HCM in relation to each disease-causing gene. Future studies should also focus on prospective follow-up of the patient to assess the disease's natural history in relation to its pathogenic variants.
CONCLUSION
This study was conducted on a group of 10 probands with HCM and their family members. A targeted Next-Generation Sequencing panel was used, and in-depth clinical profiling was carried out to study the prevalence of genetic defects and the genotype-phenotype correlation in this group of patients. This case series highlights the importance of genetic testing for clinically confirmed HCM patients and family members. The genetic information can be used as a molecular marker to complement the clinical presentation for the diagnosis of HCM, as well as a prognostic tool for the patients and their family members.
AUTHORS’ CONTRIBUTION
Hung Manh Pham, Van Khanh Tran, Trung Anh Mai, and Long Hoang Luong conceived and designed the study and performed the analysis. Thanh Van Ta and Huy Thinh Tran contributed to the development of the study’s idea and design. Hoai Thu Nguyen Thi, May Le Pham, and Chi Khanh Nguyen contributed to data collection and carried out the experiments. Minh Nhat Pham, Can Thuy Do, and Thanh Tuan Le performed analysis and finalized the results. Hung Manh Pham, Van Khanh Tran, Long Hoang Luong, and Trung Anh Mai contributed to the drafting of the manuscript. All authors have read and approved the final version for publication.
LIST OF ABBREVIATIONS
| HCM | = Hypertrophic Cardiomyopathy |
| LV | = Left Ventricular |
| ECG | = Electrocardiography |
| ESC | = European Society of Cardiology |
| SCD | = Sudden Cardiac Death |
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The study protocol was approved by the Ethics Committee of Hanoi Medical University (Hanoi Medical University Institutional Review Board, Hanoi, Vietnam, reference number: IRB00003121).
HUMAN AND ANIMAL RIGHTS
No animals were used that are the basis of this research. The study complies with the Declaration of Helsinki regarding the use of human samples and identifiable information.
CONSENT FOR PUBLICATION
The patients gave consent to publish their information, including clinical and genetics information. No identifiable information was disclosed in any form.
AVAILABILITY OF DATA AND MATERIALS
The datasets used and/or analyzed during the current study are available from the corresponding author, [T.H.T] upon reasonable request. We, however, cannot provide personal information or data containing the identification of the patients in any form.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
The authors would like to sincerely thank the patients and their parents for allowing them to publish the data. They would also like to thank their colleagues who helped them in patients recruitment and clinical data collection.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publisher’s website along with the published article.

