All published articles of this journal are available on ScienceDirect.

Role of Warburg Effect in Cardiovascular Diseases: A Potential Treatment Option
Authors Info & Affiliations
Abstract
Background:
Under normal conditions, the heart obtains ATP through the oxidation of fatty acids, glucose, and ketones. While fatty acids are the main source of energy in the heart, under certain conditions, the main source of energy shifts to glucose where pyruvate converts into lactate, to meet the energy demand. The Warburg effect is the energy shift from oxidative phosphorylation to glycolysis in the presence of oxygen. This effect is observed in tumors as well as in diseases, including cardiovascular diseases. If glycolysis is more dominant than glucose oxidation, the two pathways uncouple, contributing to the severity of the heart condition. Recently, several studies have documented changes in metabolism in several cardiovascular diseases; however, the specific mechanisms remain unclear.
Methods:
This literature review was conducted by an electronic database of Pub Med, Google Scholar, and Scopus published until 2020. Relevant papers are selected based on inclusion and exclusion criteria.
Results:
A total of 162 potentially relevant articles after the title and abstract screening were screened for full-text. Finally, 135 papers were included for the review article.
Discussion:
This review discusses the effects of alterations in glucose metabolism, particularly the Warburg effect, on cardiovascular diseases, including heart failure, atrial fibrillation, and cardiac hypertrophy.
Conclusion:
Reversing the Warburg effect could become a potential treatment option for cardiovascular diseases.
1. INTRODUCTION
The heart pumps blood throughout the body via the circulatory system [1]. This process requires energy intake from carbohydrates, lipids, and proteins. Cardiomyocytes are heart muscle cells that can convert glucose or ketone bodies into mechanical energy [2]. While cardiomyocytes predominantly use fatty acids (FAs) as their energy source, glucose provides approximately 25% of the total energy required by the heart [3, 4]. This is different from other cells where almost 70% of glucose is used as the energy source via oxidative phosphorylation; the rest is obtained via glycolysis. In the human heart, approximately 60-70% of the ATP produced in the mitochondria is used as an energy source, whereas the remainder is used for calcium homeostasis, ion pumping into the sarcoplasmic reticulum via calcium ATPase, and cell death signaling [5]. The requirement for ATP varies depending on the cell type, cellular condition, growth status, and microenvironment [6].
In conditions of impaired myocardial energy, such as cardiac hypertrophy, a major metabolic shift occurs from FA oxidation to glucose utilization, affecting glucose uptake and glycolysis rate [7]. This is accompanied by a decrease in cardiac mitochondrial oxidative metabolism and an increase in glycolysis (pyruvate conversion into lactate) [8]. The energy produced via mitochondrial glucose oxidation is greater than that produced via glycolysis [9, 10]. However, cardiac hypertrophy and Heart Failure (HF) rat models decrease myocardial glucose oxidation. Several studies have shown that changes in energy metabolism in the heart increase cardiovascular disease severity, including cardiac hypertrophy and HF [11-13].
In HF, glucose metabolism increases glucose uptake and glycolysis, ultimately leading to uncoupling of glycolysis and glucose oxidation. This may also contribute to the development of HF by increasing cardiac hypertrophy [14]. In proliferating cells, such as cancer cells, uncoupling of glycolysis and glucose oxidation occurs, which plays an important role in promoting cell growth [6, 15]. In 1920, Otto Warburg reported an increase in glycolysis and lactate production in cancer cells compared with normal cells, despite the presence of oxygen. This is known as the “Warburg effect” [16]. Although glycolysis only produces two ATP molecules compared with oxidative phosphorylation, which produces 36 ATP molecules, glycolysis is a relatively quick process [17], and the total amount of ATP produced by glycolysis is higher than that by oxidative phosphorylation in the same amount of time [18, 19]. This “Warburg” phenomenon may also occur in other conditions, such as cardiac hypertrophy, which can, in turn, result in HF because increased pyruvate dehydrogenase kinase (PDK) activity is observed, suggesting an increased rate of glycolysis [20, 21].
Recent studies have proposed the existence of the Warburg effect in cardiovascular diseases. The metabolites produced by glycolysis play an important role in regulating extracellular matrix formation, cell proliferation, apoptosis, and autophagy [22]. In addition, glycolytic enzymes are found in many diseases, including pulmonary fibrosis, hypertension, HF, atrial fibrillation (AF), and cardiac hypertrophy [23-25]. The next section focuses on the metabolic shift involved in the signaling pathways and the Warburg effect in cardiovascular disease, and discusses their potential roles in cardiovascular disease therapy.
2. MATERIALS AND METHODS
Our aim was to review the Warburg effect in cardiovascular diseases and its molecular interactions as targeted therapy. We conducted a literature search using PubMed and Google Scholar for articles published up until 2020. The keywords used were: “cardiovascular disease” or “cardiac hypertrophy” or “heart failure” or “failing heart” and “Warburg effect,” “heart metabolism” or “heart failure metabolism” or “cardiac hypertrophy metabolism” or “atrial fibrillation metabolism,” and “heart failure treatment” or “cardiac hypertrophy treatment” or “atrial fibrillation treatment.” The inclusion criteria were articles published up until 2020; those focusing on the Warburg effect in cardiovascular disease; and those regarding metabolism and treatment in cardiac hypertrophy, heart failure, and AF that were related to the mechanisms of the Warburg effect.
3. RESULTS AND DISCUSSION
The initial search found 388 article sources. After duplicates were removed, 351 records remained. A further 184 records relevant to the inclusion criteria were articles published up until 2020; those focusing on the Warburg effect in cardiovascular disease and those regarding metabolism and treatment in cardiac hypertrophy, heart failure, and AF that were related to the mechanisms of the Warburg effect. A total of 162 potentially relevant articles after the title and abstract screening were screened for full-text. Finally, 135 papers were included for the review article.
3.1. Energy Metabolism in Cardiomyocytes
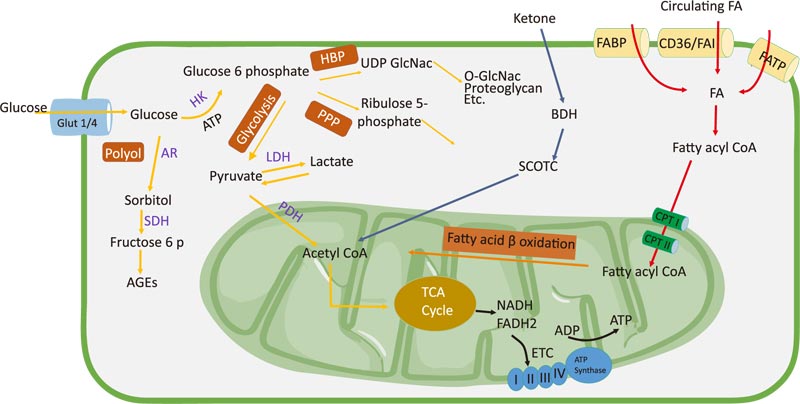
The adult heart pumping 10 tons of blood a day requires approximately 6 kg of ATP. The heart obtains energy from various metabolites, including glucose, FAs, lactate, and ketones [26]. Under normal heart conditions, >95% of the ATP is produced via oxidative phosphorylation; the rest is obtained via glycolysis. Approximately 50-70% of the ATP is produced via the oxidation of free FAs [27, 28].
The transport of FAs from the plasma to cardiomyocytes is facilitated by fatty acid translocase (FAT/CD36), fatty acid transport protein, plasma membrane fatty acid-binding protein (FABPpm), and heart-type fatty acid-binding protein [29]. FAs are esterified to fatty acyl-CoA, which enters the mitochondrial membrane with the help of carnitine palmitoyltransferase I and II (CPT I and II). Fatty acyl-CoA is converted to acetyl-CoA, which then enters the tricarboxylic acid (TCA) cycle to produce nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2). These cofactors are used in the electron transport chain to produce ATP [30]. Malonyl-CoA negatively regulates CPT I activity and is formed via acetyl-CoA carboxylase (ACC) and degraded by malonyl-CoA decarboxylase. AMP-activated protein kinase (AMPK) phosphorylates ACC, which inhibits malonyl-CoA formation and increases FA oxidation. Under normal heart conditions, FA oxidation, esterification, and uptake are activated by peroxisome proliferator-activated receptor coactivator-1α (PGC1α), which stimulates peroxisome proliferator-activated receptor α (PPARα). PPARα can modify glucose oxidation by inhibiting pyruvate dehydrogenase (PDH) via pyruvate dehydrogenase kinase 4 (PDK4) [31]. PDK is activated by NADH and acetyl-CoA, which are produced via FA oxidation, and directly inhibits PDH. PDK is inhibited by pyruvate and reduced the acetyl-CoA/free CoA and NADH/NAD+ ratios [32].
The main sources of glucose used by the heart are via the uptake of exogenously obtained glucose and glycogen stored in the liver. Glycolysis plays a critical role in cell metabolism and produces pyruvate, NADH, and ATP. Pyruvate is converted to lactate by lactate dehydrogenase (LDH) and mostly undergoes mitochondrial glucose oxidation [33]. Glucose from the circulation is transported to cells via glucose transporters, including GLUT1 and GLUT4. In the fetal heart, GLUT1 is the major glucose transporter, and its level increases in response to myocardial stress or injury. In contrast, GLUT4 is the predominant isoform in the adult heart and plays a role in basal myocardial glucose uptake [34]. The deletion of GLUT4 in mice hearts leads to metabolic adaptations [35]. GLUT 4-knockout mice (G4H−/−) show changes in glycolysis as a defense mechanism against ischemic injury [36]. After being absorbed in the intestine, glucose is phosphorylated to form glucose-6-phosphate (G6P), which then enters the metabolic pathway. In the mitochondria, pyruvate is decarboxylated by pyruvate dehydrogenase to form acetyl-CoA, which then enters the TCA cycle to produce NADH and FADH2. Pyruvate oxidation is regulated by PDH. ATP plays an important role in calcium absorption in the sarcoplasm and relaxation during diastole [37]. Glucose is catalyzed by glycolysis and intermediate glycolysis. Glucose is converted to G6P by hexokinase (HK). G6P then enters the pentose phosphate pathway (PPP) [38]. PPP produces NADPH, which maintains levels of reduced glutathione and ribulose 5-phosphate used for nucleotide biosynthesis or for the generation of intermediates for glycolysis [39, 40]. The polyol pathway is another glycolytic pathway in which glucose is converted to sorbitol by aldose reductase (AR). Sorbitol dehydrogenase then oxidizes sorbitol to fructose and advanced glycation end-products [41]. A study has reported that AR activity increased in the heart and kidneys of a hyperglycemic mouse model [42]. The hexosamine biosynthetic pathway generates the end products hyaluronan, glycosylphosphatidylinositol anchor, glycolipid, proteoglycan, N-glycan, and O-linked β-N-acetylglucosamine (O-GlcNAc), which are derived from uridine diphosphate N-acetylglucosamine [33]. In cardiac hypertrophy, inhibition of O-GlcNAc can reduce the progression of the development of hypertrophy [43, 44].
Ketone bodies, such as acetoacetate and β-hydroxybutyrate, are energy sources produced via FA β-oxidation in the liver [45]. Ketones are oxidized in the heart, brain, and skeletal muscle [30]. During terminal oxidation, ketones are converted back to acetyl-CoA in a reaction catalyzed by succinyl-CoA-3-oxoacid-CoA transferase, β-hydroxybutyrate dehydrogenase 1, and acetyl-CoA acetyltransferase 1-or through the lipogenic pathway via fatty acid synthase [46]. The heart uses ketones to maintain myocardial function in HF. In HF, the decrease in FA oxidation makes ketone bodies an efficient fuel for ATP production [47] (Fig. 1).
3.2. Warburg Effect in Cardiovascular Diseases
Under normal heart conditions, FAs are the main source of ATP production. In contrast, under various pathological conditions, including cardiac hypertrophy, there is a shift in energy metabolism from mitochondrial oxidative phosphorylation and the TCA cycle to glycolysis [48]. The mass changes in cardiac hypertrophy are associated with metabolic changes, resulting in switching to more ATP production from glucose than from FAs. Glycolysis is a biochemical process that converts glucose to pyruvate and then to lactate and CO2 to produce ATP. Carbon atoms obtained from glucose are either derived exogenously or endogenously by glycogen breakdown [49]. The change in the rates of glycolysis and oxidative phosphorylation results in the uncoupling of glycolysis and glucose oxidation, which interferes with cardiac function [8]. In HF, there is an increase in glycolysis with or without a decrease in mitochondrial oxidation [20, 50]. Impaired myocardial energy metabolism in cardiac hypertrophy is characterized by increased glucose demand and decreased FA oxidation. These alterations are associated with increased glycolysis in cardiac hypertrophy [51-54]. Previous studies have shown that an increase in glucose uptake and the activation of phosphofructokinase (PFK), play a role in increasing the rate of glycolysis in rats with hypertrophied hearts. In left ventricular hypertrophy (LVH), there is an increase in adenosine monophosphate (AMP) and adenosine diphosphate (ADP), triggering AMPK, which in turn increases glucose transport and PFK activation [54].
Dahl salt-sensitive rats with HF and preserved ejection fraction showed decreased FA metabolism and increased glucose metabolism. Uncoupling between glycolysis and glucose oxidation, resulting from increased glycolysis, occurs without changes in glucose oxidation; this results in an acidic environment that hampers cardiac function [8]. The Warburg effect is known to occur in cancer cells. These cells have high proliferation rates and exhibit high glycolysis rates in the presence of oxygen [55, 56]. In addition to cancer cells, the Warburg effect may occur in other cells with high energy demand. This effect has been demonstrated in cardiac hypertrophy, which causes HF. Inhibition of anaerobic glycolysis can reduce hypertrophy and increase the flexibility of cardiac metabolism to maintain the need for cardiac output to meet the high metabolic demand [57]. HF and hypertrophy mouse models showed decreased myocardial glucose oxidation levels. Metabolic changes are believed to increase the severity of HF [30].
Waleed et al. developed a post-infarction mouse model and revealed an imbalance in glucose metabolism and increased proton production, resulting in the deterioration and disruption in mechanical left ventricular function [14]. Systolic dysfunction and glucose oxidation damage in HF may be caused by mitochondrial dysfunction, decrease in the expression of the genes involved in glucose oxidation, or an increase in the expression of the PDH complex [11, 58]. Increased oxidative stress may be a cause of mitochondrial dysfunction in cardiac hypertrophy, HF, and AF. Reactive oxygen species (ROS) play a role in cardiac remodeling in the stress response via increased mitochondrial biogenesis, cardiomyocyte volume, and capillary density [59]. During chronic stress, ATP demand increases in the heart, leading to increased activity of the electron transport chain in the mitochondria and ROS generation. This results in a shift in the energy metabolism to glycolysis, even in the conditions of adequate oxygen levels, as compensation for the high energy demand. This could be the underlying mechanism of the Warburg effect in cardiovascular diseases, particularly HF.
Other studies have shown that FA oxidation stimulation and oxidative phosphorylation improve HF [11, 14, 60]. We previously reported that mice with CD36 genetic deletions showed decreased FA uptake and increased glycolysis flux; this, in turn, increased the progression from compensated hypertrophy to HF. This phenomenon was also observed in mice models of heart pressure overload with a double knockout of FABP4 and FABP5, with increased glycolysis and decreased FA uptake, resulting in severe hypertrophy, fibrosis, and cardiac dysfunction. Furthermore, the use of glucose was demonstrated not only for catabolic pathways but also for anabolic pathways to synthesize biomass [61, 62]. CD36 increased the expression of Forkhead Box O1 and PDK4 via the activation of PPARδ/β to inhibit glucose oxidation [63]. This could support the association between the Warburg effect and cardiovascular diseases due to increased glycolysis, even accompanied by glucose oxidation. The increase in glycolysis in this study [61-63] may have involved the Warburg effect, although it remains unclear. Further research and clinical studies in humans are needed.
Structural and electrical remodeling also occurs in AF [64]. In acute AF, contraction and electrical activity increase four- to six-fold compared with normal atrial conditions. Tu et al. reported the downregulation of FA metabolism in the atria of permanent AF due to the downregulation of the enzymes regulating FA oxidation [65]. Tsuboi et al. reported that the deletion of mitochondrial DNA in patients with AF led to the disruption of ATP synthesis [66]. Decreased oxidation of pyruvate and FAs increases glycolysis in AF, leading to the upregulation of glycolytic enzymes, lactate production, and the downregulation of the PDH complex [25, 37]. Myocardial fibrosis and metabolic stress are related to the Warburg effect and play a role in the development of AF. An increase in PDK4 is associated with an increase in myocardial fibrosis.
The Warburg theory suggests an ATP production shift from the TCA cycle and oxidative phosphorylation to glycolysis or lactate production, even in the presence of oxygen, and is thus termed aerobic glycolysis [55, 67, 68]. Under normal conditions, most of the glucose taken up by cells is metabolized via glycolysis to pyruvate, which then enters the TCA cycle and electron transport chain to generate ATP via oxidative phosphorylation [69]. Studies have shown that the increase in oxidative phosphorylation in cancer cells can be achieved by inhibiting glycolysis. Activation of glycolysis in the proliferative cells supports the cells to grow [15, 70]. Glycolysis can be induced by hypoxia via activation of hypoxia-inducible transcription factor 1α (HIF-1α). HIF-1 is a heterodimeric transcription factor that changes hypoxic gene expression and consists of HIF-1α and HIF-1β subunits. It is thought to be adaptive for cells exposed to an oxygen-deficient environment. PDK1 is the target of the HIF-1α gene, which increases PDK activation and leads to the inhibition of PDH, which allows pyruvate to enter the mitochondria to perform oxidative phosphorylation. PDK1 regulates mitochondrial function in hypoxic states by inhibiting the entry of pyruvate into the TCA cycle, indirectly inhibiting PDH [71-74].
In a monocrotaline-induced model of pulmonary hypertension, the glycolysis rate of right ventricular hypertrophy (RVH) was increased compared with that of the control group, and there was no increase in glucose oxidation. An increase in GLUT1 was observed as a compensatory mechanism of low ATP levels. Decreased glucose oxidation is thought to occur due to PDH phosphorylation [20]. Decreased glucose oxidation rate in fawn-hooded rats with pulmonary hypertension and RVH occurs due to increased PDK4 levels. Therefore, inhibition of PDK can increase PDH activity to prevent HF [21]. Kato et al. reported metabolic remodeling in compensated left ventricle hypertrophy and HF and showed an increase in glycolysis, characterized by an increase in lactate and a decrease in PDH [11]. Studies have reported that an increase in LDH, which converts pyruvate to lactate, results in increased lactic efflux in the hypertrophied heart muscle [75]. PH alteration and increase in protons can impair the sensitivity of troponin I to calcium, which interferes with cardiac contractility [76]. The upregulation of glycolysis rates results in uncoupling between glycolysis and glucose oxidation, leading to an increase in proton and lactate production [77, 78]. This suggests that an increase in lactate levels is due to a higher glycolysis rate compared with mitochondrial oxidation and that increased PDK inhibits the activation of the PDH complex and the biosynthesis of the precursors, which are needed to be required for the development of hypertrophy [23, 79]. Decreased oxidation of pyruvate and FAs increases glycolysis in AF, leading to the upregulation of glycolytic enzymes, lactate production, and the downregulation of the PDH complex. The Warburg effect increased glucose use and fibrosis levels in a canine model of p-AF cardiomyopathy, which is characterized by the increased expression of PDK1, PDK4, LDHA, and lactic acid and decreased expression of isocitrate dehydrogenase and PDH [80]. Increased glycolytic enzymes in cardiovascular diseases, as seen in cancer cells, indicate the involvement of the Warburg effect.
HIF-1α plays an important role in energy changes from oxidative phosphorylation to glycolysis. HIF-1α is the main hallmark and regulator of the Warburg effect in cancer. It is upregulated under cardiac hypertrophy conditions [79, 81, 82]. Increased HIF-1α expression in AF is an early response of atrial cardiomyocytes. In AF models, there was a decrease in PPARα/PGC1α activity that was inhibited by HIF-1α, which caused a decrease in FA metabolism [81, 83]. HIF-1α plays a role in metabolic reprogramming in end-stage HF. Furthermore, it regulates the gene expression of PDK1 and LDHA, which play a role in reprogramming glucose metabolism [84]. Interactions between HIF-1α and pyruvate kinase M2 (PKM2) in the nucleus activate the transcription of genes involved in glycolysis, such as GLUT 1, LDHA, and PDK1 [85]. PKM2 is considered the hallmark of the Warburg effect in cancer cells [86]. HIF-1α activates the conversion of adult pyruvate kinase M1 (PKM1) to fetal PKM2. PKM1 is normally expressed continuously in normal cells, whereas PKM2 is widely expressed in cancer cells [87]. Metabolic changes that occur in heart diseases cause a metabolic switch back to fetal metabolism, in which the glycolysis rate exceeds that of oxidative phosphorylation [88, 89]. The fetal heart uses glucose as the main energy source. A few days after birth, the heart switches to using FA oxidation as its main energy source. In the heart of a newborn rabbit, approximately 44% of the ATP was derived via glycolysis; however, after 7 days, only approximately 7% of the ATP was produced via glycolysis [87, 90, 91] In cancer cells, HIF-1 suppresses the TCA cycle via PDK activation and functions to maintain ATP and ROS production [72]. The increase of PDK regulation leads to reduced levels of active PDH via PDH phosphorylation [73]. Suppression of HIF-1α prevents hypertrophy-induced cardiac dysfunction [92]. The Warburg effect and HIF-1α are thought to result in the metabolic shift to glycolysis in both cancer cells and cardiovascular diseases (Table 1).
| No. | Disease | Model | Event | Reference |
|---|---|---|---|---|
| 1 | LVH | Weanling male Wistar rats | Glycolysis ↑ Lactate ↑ |
[54] |
| 2 | LVH HF |
Inbred male Dahl Salt-sensitive rats | Glycolysis ↑↑ Lactate ↑ |
[11] |
| 3 | LVH | Male Wistar rats | PDH ↓ PDK ↑ |
[93] |
| 4 | HF | Male C57BL/6 mice with coronary artery ligation or sham operation procedure | Glycolysis ↑ Glucose oxidation ↑ Lactate ↑ |
[14] |
| 5 | HF | Male Dahl salt-sensitive rats with induced HfpEF | Glycolysis ↑ Glucose oxidation ↔ FA oxidation ↓ Lactate ↑ Proton ↑ |
[8] |
| 6 | LVH | Mb transgenic mice with transverse aortic constriction surgery | Glycolysis ↑ F-2,6-P2 ↑ |
[94] |
| 7 | RVH | Adult male Sprague–Dawley rats injected monocrotaline | Glycolysis ↑ Glucose oxidation ↔ PDH ↓ PDK ↑ |
[20] |
| 8 | RVH | Fawn-hooded rats | Glycolysis ↑ Glucose oxidation ↓ PDK ↑ |
- |
| 9 | Chronic cardiac hypertrophy | Mouse transaortic-constriction | Glycolysis ↑ p-PDH ↑ |
[95] |
| 10 | AF | Beagle Canine model of paroxysmal atrial fibrillation | Glycolysis ↑ LDHA↑ PDK1 ↑ PDK4 ↑ |
[80] |
3.3. Warburg Effect: Targeted Treatment in Cardiovascular Diseases?
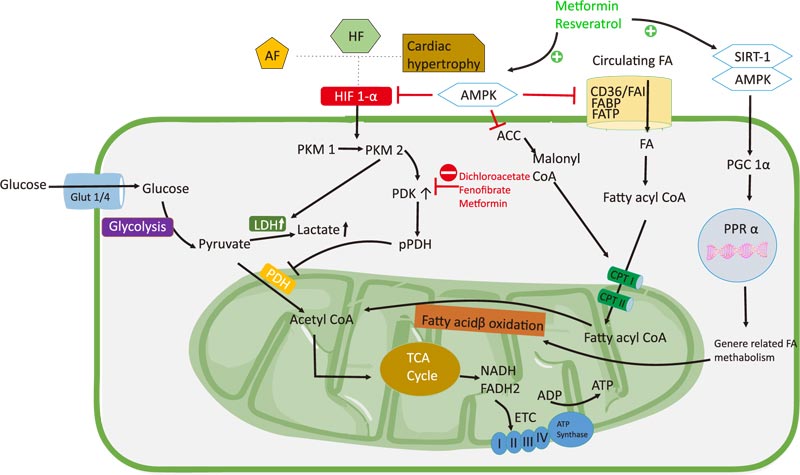
In cardiac hypertrophy, cell growth and proliferation are accompanied by decreased FA β-oxidation, which is characterized by changes in the expression of enzymes involved in glycolysis and FA β-oxidation [33]. In HF, ATP is decreased to 30-40% compared with that in the normal heart due to a switch to the use of glucose as the primary energy source instead of FAs. In HF, a decrease in myocardial ATP up to 60-70% is accompanied by a decrease in mitochondrial glucose oxidation, increased glycolysis, and decreased creatine kinase activity [11, 30, 96]. Therefore, promoting the coupling of glucose oxidation and glycolysis is one approach to treat HF because it enhances oxidation [97]. Correcting cardiac function via the reverse Warburg effect can be achieved by stimulating glucose oxidation [8].
Dichloroacetate (DCA) can induce oxidative phosphorylation and the activation of the PDH complex and has been widely studied. Inhibition of aerobic glycolysis increases cardiac metabolic flexibility and reduces hypertrophy via PDH inhibition. In ischemia-reperfusion models, DCA has a cardioprotective mechanism [98, 99]. In an MCT-RVF, RVH leads to an increase in PDK activity. The administration of DCA, a pyruvate dehydrogenase kinase inhibitor, decreased the expression of PDK in MCT-RVF mice [20]. DCA decreased LDHA and upregulated superoxide dismutase 2 [100]. In Dahl-sensitive rats, the use of DCA treatment prevented the progression of LVH to HF. DCA was shown to improve cardiac function and survival in the rat model [11]. Interestingly, DCA treatment in mice with hyperthyroidism showed an increase in PDH activity and increased myocardial flexibility and reduced hypertrophy [93]. Clinical data on the use of DCA remains sparse. A clinical study of 10 patients with HF (New York Heart Association class III and IV) infused with DCA intravenously showed improved LV stroke volume and myocardial oxygen consumption [101]. The effect of DCA might inhibit the Warburg effect by improving the coupling of glycolysis to glucose oxidation by inhibiting PDK to activate PDH, thereby increasing pyruvate oxidation and mechanical efficiency.
Increased AMPK activity is associated with cardiac hypertrophy and HF. Targeting AMPK activity as a treatment in HF models improved cardiac function and reduced ventricular remodeling [102, 103]. AMPK phosphorylation is increased in patients with paroxysmal AF as an adaptive response to metabolic stress in AF for Ca2+ handling and contractility [57]. Furthermore, AMPK increased levels of FA transport proteins FAT/CD36 and FABPpm [104]. In a mouse AF model, AMPK was associated with an increase in FAT/CD36, leading to excess uptake of FAs and decreased expression of GLUT4 in the membrane. Therefore, AMPK has cardioprotective functions and prevents the AF progression by increasing FA oxidation compared with glucose oxidation [51, 105]. Some drugs, such as metformin, statins, and resveratrol, activate AMPK. Metformin increases AMPK activity by directly increasing its phosphorylation or binding to AMPK subunits, thereby increasing the heterotrimeric complex. Metformin indirectly changes the ratio of AMP or ADP to ATP [106, 107]. In animal models, metformin phosphorylates AMPK and increases nitric oxide levels, thereby improving cardiac function [108]. The use of metformin as an AMPK activator in a Beagle dog AF model showed an increase in AMPK, PGC1α, and PPAR1α activity, and a decrease in HIF1α, GLUT1, PDK, HK, and LDH. Metformin treatment also revealed that an increase in PDH is the key to the Warburg effect [109]. A clinical study has shown that the use of metformin reduces the mortality rate in ambulatory HF patients with diabetes [110]. AMPK activates FA metabolism via PGC1α/PPAR1α and inhibits HIF-1α to reverse the Warburg effect [25]. Resveratrol is a natural polyphenol found in plants, including peanuts, cranberries, and grapes. It has a cardioprotective effect via the activation of AMPK/sirtuin 1, which is involved in signaling and modulating cardiac metabolism. The beneficial effects of resveratrol in HF have been shown in ischemic and non-ischemic HF animal models [111-113].
The activation of PPARα and PGC1α increases FA flux in cells as well as increases the activity of enzymes that play a role in FA β-oxidation [114]. Mice lacking PPARα were shown to have increased glycolysis and decreased FA oxidation, characterized by a decrease in protein transport and in enzymes playing a role in β-oxidation (such as malonyl-CoA decarboxylase, which inhibits CPT I) [115]. In mouse HF models, treatment with fenofibrate, a PPARα agonist, led to increased FA metabolism [116, 117] as well as the upregulation of PDH, GLUT4, PPARα, PGC1α, and p-sirtuin 1 and downregulation of PDK4 [118]. In dogs with high-frequency left ventricular pacing, fenofibrate administration prevented a decrease in FA metabolism and improved cardiac function [119]. The reverse Warburg effect can be generated by promoting the coupling of glycolysis and glucose oxidation and can be achieved by enhancing glucose and FA oxidation. Metabolic therapy by disrupting the Warburg effect represents a potential treatment for cardiovascular diseases, although more extensive human studies are warranted to demonstrate this effect as well as the clinical implications of the Warburg effect on cardiovascular diseases (Fig. 2).
3.4. Dietary and Exercise Modifications in Cardiovascular Diseases
Behavioral modifications, such as a healthy lifestyle, including healthy dietary patterns and appropriate exercise, are important strategies recommended for the prevention of cardiovascular disease [120]. In recent years, epigenetic studies have shown that environmental factors, including diet and physical exercise, may change the epigenome.
Meta-analyses and randomized controlled trials have shown that a Mediterranean diet consisting of fruits, grains, vegetables, nuts, fish, unsaturated fats, and legumes reduces mortality and morbidity in patients with cardiovascular diseases [121, 122]. This healthy diet pattern should be low in trans fats, refined grains, red and processed meat, sugary drinks, and added sugars [123]. Cohort studies have shown an association between metabolite profiles and high adherence to dietary recommendations proposed by the Alternative Healthy Eating Index (AHEI) in patients with cardiovascular diseases [124]. High levels of monounsaturated and saturated FAs are associated with a high risk of cardiovascular diseases. Furthermore, a high ratio of omega-3 polyunsaturated fatty acids (PUFAs), such as docosahexaenoic acid, to total FAs is associated with a reduced risk of cardiovascular diseases [124]. A hereditary cardiomyopathy model demonstrated that omega-3 PUFAs can reactivate the expression of genes damaged in cardiomyocytes and also prevent plasma membrane degradation, which maintains signal transmission from the membrane surface to the nucleus [125]. Nutrition can modulate myocardial genes by activating the transcription factor PPARα [126, 127]. A population-based prospective cohort study has found that dietary intake of different PUFAs alters the association between genetic variation in fatty acid desaturase and high-density lipoprotein and low-density lipoprotein (LDL) [128]. Reduced risk of cardiovascular diseases is associated with the intake of polyphenol-rich diets related to DNA methylation. Polyphenols are present in fruits and vegetables, including cocoa and green tea. In vitro studies have shown that cocoa extract can inhibit the expression of genes encoding DNA methyltransferases and methylenetetrahydrofolate reductase, which are found in several disease conditions, including heart diseases [129, 130]. Cocoa polyphenol is cardioprotective and is associated with decreased LDL levels. Decreased LDL levels can lead to a reduced risk of atherosclerosis leading to cardiovascular disease [131, 132]. Dietary folic acid and vitamin B may reduce the risk of cardiovascular disease via the methylation of homocysteine to methionine, and folic acid deficiency increases cardiovascular diseases via DNA methylation [133].
Mouse model studies have shown increased expression of regulators of lipid metabolism, such as CPT I, PPARα, and sterol regulatory element-binding protein 1c, and the upregulation of CD36 gene expression [134, 135]. Dietary modifications and appropriate exercise can contribute to genetic changes in cardiovascular metabolism via epigenetics.
CONCLUSION
The Warburg effect is an energy shift in glucose metabolism from oxidative phosphorylation to aerobic glycolysis that occurs in cancer as well as other non-cancer conditions, including cardiovascular diseases. The Warburg effect may also occur in highly proliferative cells involved in cardiovascular diseases such as cardiac hypertrophy, HF, and AF. The increased activity of glycolytic enzymes in cardiovascular diseases, as observed in cancer cells, indicates the involvement of the Warburg effect. The mechanism of the Warburg effect in the heart is not completely understood, although HIF-1α is thought to play a role in its regulation. The Warburg effect can be used as a therapeutic target for improving heart conditions by inhibiting the shift of glucose oxidation to aerobic glycolysis. This can be achieved by increasing glucose oxidation via glycolysis inhibitors and increasing FA oxidation. Diet and exercise can also be used to prevent cardiovascular diseases. Further clinical studies are warranted to determine the involvement of the Warburg effect in cardiovascular diseases, identify more therapeutic targets, and assess the effect of epigenetics on the Warburg effect in cardiovascular diseases.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
This study was funded by the internal research grant of Universitas Padjadjaran and Universitas Pasundan Indonesia with grant number 16/Unpas.R/G.1/I/2020.
CONFLICT OF INTEREST
The authors confirm they have no conflicts of interest, financially or otherwise.
ACKNOWLEDGEMENTS
This study was supported by Universitas Padjadjaran and Universitas Pasundan.

