All published articles of this journal are available on ScienceDirect.

Nitric Oxide and Related Aspects Underlying Angina
Abstract
Increased number of patients affected by metabolic syndrome (MS) has prompted the necessity of better understanding what is involved in such syndrome. Nevertheless, the establishment of promising therapies depends on the knowledge about the interaction of molecules within MS. In such context, Nitric Oxide (NO) emerges from a bulk of works relating its roles on aspects of MS, including cardiovascular diseases, their symptoms and comorbidities, which are thought to be triggered by similar sources. NO, nitric oxide synthase and enzymatic chains are keys for those disease and symptoms processes. NO has been separately described as part of hypertensive, ischemic and pain signaling. Although there are similar pathways likely shared for generating cardiovascular symptoms such angina, they are barely associated to NO in literature. The present review aims to clarify the patterns of NO alteration in metabolic syndrome directly concerned to cardiovascular symptoms, especially angina.
1. INTRODUCTION
Angina is the prominent symptom of coronary heart disease (CHD), a condition, among others that describes one of the main disorders related to metabolic syndrome (MS). This syndrome is accepted to be an association among obesity, hypertension, dyslipidemia, glucose metabolism alteration (glucose intolerance, insulin resistance or diabetes type II) and responsible for the higher risk of cardiovascular diseases [1]. Studies around the world have been observing individuals with MS presenting inferior prognosis and sharp mortality when comparing with non-MS patients [2-6].
Multicenter studies have demonstrated the Nitric Oxide (NO) influences in cardiovascular system. Even though hypertension, vascular diseases and metabolic syndrome have been related to important symptom such as angina, the absence or deviations in NO signaling are scarcely related to cardiovascular disease and/or their symptoms. The importance of this association consists of NO controlling relevant functions such as neurotransmission [7, 8], vascular tone [9, 10], gene transcription [9, 10], mRNA translation [11, 12] and post-translational modifications of proteins [13, 14]. Overall, despite the lacking of association in literature, NO plays a trivial role in angina.
The main damages involved on the NO signaling are related to oxidative stress and the development of the components predisposing MS and its symptoms. In the present review, we sought to clarify the patterns of NO alteration in metabolic syndrome directly concerned to cardiovascular symptoms, especially angina.
2. THE GENERATION OF NITRIC OXIDE
NO is produced from dietary sources via Nitrate-Nitrite-NO pathway or from endogenous turnover. The main differences between these pathways are basically the enzymes, the substrates, respectively nitrate and L-arginine, and the requirement of molecular oxygen (O2) for NO turnover pathway. Even though there are two processes, they are linked by the reduction of endogenously produced NO, which provides the largest endocrine source of directly bioavailable NO to inorganic nitrite (NO2−) [15].
The NO synthesis from dietary intake is dependent on xanthine oxidoreductase (XOR). Among other functions, XOR is a major NO2− reductase enzyme linked to cellular NO signaling events [16-33] Fig. (1). This enzyme is essential for nitrate (NO3−) use from diet.
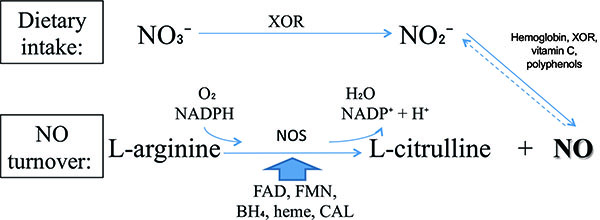
In other way, NO produced from L-arginine requires an enzyme called nitric oxide synthase (NOS). There are three classical isoforms: endothelial (eNOS), neuronal (nNOS) and inducible (iNOS). These isoenzymes are derived from different genes and trigger diverse organic processes [34]. eNOS and nNOS are constitutively expressed and are dependent on Ca2+ for activation. In contrast, iNOS is usually expressed in proinflammatory processes and Ca2+ independent [35-43]. Besides the classical, there is a novel NOS isoform, mitochondrial NOS (mtNOS), which is present in mitochondria [44-46] and appears to regulate cellular oxygen consumption/energy metabolism without engendering oxidative stress [47, 48]. Positive vascular effects are well established as mediated by cellular pathways of NOS/L-arginine NO signaling [35, 49].
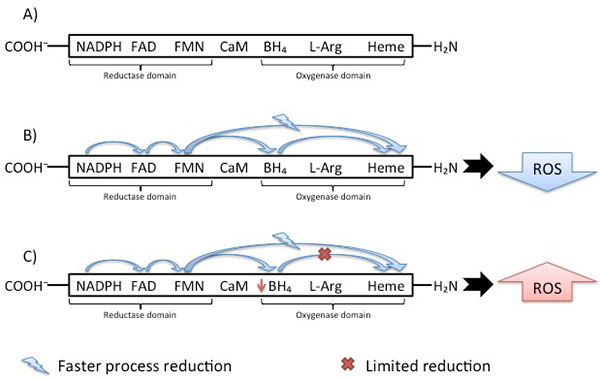
As almost all enzymes, NOS isoforms require cofactors. Tetrahydrobiopterin (BH4) is one of the critical cofactors for NOS activity. In conditions that will be approached in this article such as hypertension, BH4 is oxidized leading to NOS uncoupling [50, 51], increased Reactive Oxygen Species (ROS) and reduced NO production due to an electron flowing through the enzyme (Fig. 2).
3. NITRIC OXIDE INFLUENCES
NO acts on a number of protein targets through cell signaling. One of the most important physiological signaling is the activation of soluble guanylyl cyclase (GC) and the generation of cyclic guanosine monophosphate (cGMP) [9, 10, 52-55], especially for neurotransmission and vascular tonus functions. The transduction for the NO signaling is given by its reaction with superoxide anion (O2−•), resulting in NO inactivation and potent oxidant peroxynitrite (ONOO−) formation. This compound causes oxidative damage, nitration, and S-nitrosylation of biomolecules including protein, lipids, and DNA [56, 57]. These damages are primordial to the development of the components predisposing MS and its symptoms, causing respectively hypertension and pain.
3.1. NO Pathway in Pain
The cardiovascular system functions reported on this article relies on blood circulation; namely, the association of angina and hypertension regarding nervous systems and blood circulation influenced by NO. In this sense, the expected NO functioning in the sensory perception of pain signaling and explanation are based on biochemical processes depicted on (Fig. 3).
The integrity of nervous system is essential for pain protective functions. Processes responsible for converting sensory stimuli to cellular transduction, enabling the recognition and characterization of the signal, modulate the frequency, rate and extent of the sensory perception of pain. Together with the NOS isoforms, glutamate receptor is involved in some of the important signaling processes [58]. Even though these proteins are reported to be associated to the regulation of sensory perception of pain, it is still unlinked, according to resources for “protein” search at the National Center for Biotechnology Information (NCBI) database [59], if they are positive or negative regulators or even part of the modulation of pain.
The usual NO pain pathway is showed on Fig. (3). However, how the NOS isoforms, mainly nNOS, are related to heart pain and the mechanism by which NO can sensitize the neuronal path to trigger the perception of pain remain partially unknown. Visceral and neuropathic are the main types of pain correlated to NO and ischemia, the principal cause of heart pain. Both pathways are nociceptors-sensitized on primary afferent C fibers, where the action potential is conducted to the central nervous and to secondary afferent neurons in spinal-cord dorsal horn. Then, the signal reaches areas of the brain responsible for localization and emotional aspects of pain, respectively, through spinothalamic and spinoreticular tracts. The main difference between visceral and neuropathic pain resides on the type of stimuli for which they respond to. Smooth muscle distension or contraction, capsule stretching surrounding an organ, ischemia, necrosis or inflammatory mediators trigger visceral pain; dissimilarly, the triggers for neuropathic pain pathway are trauma, surgery, diabetes mellitus, chemotherapy, radiotherapy, infection, malignancy and ischemia in which the damage occurs directly to central or peripheral nervous system [60].
Excluding nociception stimuli from extra cardiac issues, the main etiology for angina is ischemia. Intermittent ischemia in focal myocardial regions might result into functional alterations for both efferent and afferent cardiac adrenergic, and possibly vagal, nerve fibers. Additional mechanisms such as metabolic abnormalities might also adversely affect cardiac nerve fiber function [61, 62]. Cardiac stimuli are usually unable to elicit a painful response through afferent nerve fibers due to their low-sensitiveness; however, the fibers sensitivity to cardiac stimuli is increased if there are functional alterations, such the ones caused by ischemia. Therefore, the result is a painful response and consequent greater cardiac pain perception. This process is similar to cutaneous hyperalgesia due to peripheral sympathetic fiber injury described in literature [63]. Overall, impaired myocardial circulation generates ischemia stimulating the nociceptive pathway.
In order to elucidate the relationship between NO and angina, there are numerous studies with pharmacological approaches based on biotechnology researches applying knockout NOS mice [64-82]. Several studies have related drugs based on NO mechanisms and their influences on pain or ischemic signals.

In this sense, the development of NOS inhibitors was one of the first pharmacological approaches. Regarded as a therapy, since chronic pain patients showed a significant increase in NO plasma levels in comparison with healthy individuals [83], methylene blue (MB) is the most studied drug affecting NO mechanisms [64-73, 82]. MB directly inhibits constitutive and inducible NOS [65] through cGMP accumulation avoidance by GC enzyme blockage [65, 66]. A valuable property of MB is its antioxidant effects [66]; it acts inhibiting the formation of free oxygen radicals and O2−• by competing with molecular oxygen (O2). Therefore, the transfer of electrons by xanthine oxidase (XO) [68] is prevented. Studies have demonstrated MB decreasing pain levels in patients with chronic therapy-resistant neuropathic pain on the first 2 days after administration [69, 74].
Complementarily, studies using knockout mice analyzed NOS absence. In 2008, Nakata et al. [75] demonstrated NOS isoforms knockout mice in conditions of hypertension, hyperlipidemia, impaired glucose tolerance, insulin resistance, metabolic syndrome and presence of visceral obesity. In fact, targeted disruption of NOS genes leads to mutant mice development and allows a better understanding of NO mechanisms related to blood pressure regulation, endothelial dysfunction, response to vascular injury, response to stroke and cerebral ischemia, diet-induced atherosclerosis and cardiac contractility [76]. Results from such researches have shown the deletion of the eNOS gene led to increase blood pressure [84, 85]. Other studies analyzed the phenotype of nNOS knockout mice and noticed stomachs enlargement, several times bigger than normal size, demonstrating nNOS role in smooth muscle relaxation of the pyloric sphincter. nNOS knockout mice were also resistant to focal and global cerebral ischemia, consistent as a part of nNOS-derived NO function in cellular ischemic injury [76-80]. nNOS gene deletion has also been associated with more severe left ventricular remodeling after myocardial infarction [81].
3.2. NO Influences in Hypertension and Angina
According to data from the World Health Organization (WHO), cardiovascular diseases killed 17.5 million people in 2012, which are 3 in every 10 deaths globally distributed. Of these, 7.4 million people died due to ischemia and 1.1 million due to hypertensive heart diseases [86]. Both, ischemic and hypertensive heart diseases are directly influenced by coronary heart dysfunctions, which causes their usual symptom: angina [87]. In addition, one of the main prescriptions to patients with angina is glyceryl trinitrate, which belongs to the nitrates chemical group responsible for vasodilatation and consequent blood pressure decreasing.
There are many evidences on literature about the roles of NOS and cytochrome C (Cyt-C) in cardiovascular diseases [36, 75, 88-92]. The NOS influences on blood pressure and circulation vary depending on the type of the isoform. Nonetheless, Cyt-C is known to be part of NO production in strictly hypoxic conditions, such as ischemic angina. Despite these two well-studied proteins, there is a lack of evidence about what other factors are involved in such disrupted cardiovascular systems. In summary, it is possible only to correlate Cyt-C and NOS as part of the NO role in hypertension and angina.
The three NOS isoforms are active on cardiovascular system; however, the main enzymes related to hypertension and angina are eNOS and nNOS. iNOS contributes mostly to the pathophysiology of inflammatory diseases and septic shock [55, 75, 93].
NO produced by nNOS in nitrergic nerves is considered as a neurotransmitter responsible for stimulating NO-sensitive GC in its effector cells, thereby decreasing the tone of various types of smooth muscle including blood vessels [55, 94, 95]. nNOS functions include synaptic plasticity in the central nervous system (CNS), central regulation of blood pressure, smooth muscle relaxation and vasodilatation via peripheral nitrergic nerves [55]. Most importantly, nNOS plays a role in the regulation of vascular tone independent of effects from nNOS in the CNS [94, 95]. The blockage of nNOS activity in the medulla and hypothalamus causes systemic hypertension [96].
Complementarily, eNOS-derived NO dilates all types of blood vessels by stimulating soluble GC and increasing cGMP in smooth muscle cells [9, 10, 84]. NO from eNOS is a homeostatic regulator keeping blood vessels dilated, blood pressure, vasoprotection and anti-atherosclerotic effects. Although mostly expressed in endothelial cells, eNOS has also been detected in cardiomyocytes [94, 95]. Pharmacologically, vascular oxidative stress can be reduced and eNOS functionality restored with both renin- and angiotensin II- inhibitors and AT1 receptor blockers, and also with statins [55]. There are eNOS stimulators, the classic class of drugs for treating hypertension and myocardial infarction [35, 55, 97]. This choice for treatment is due to the powerful protective effect of eNOS-derived NO against the onset of atherogenesis. In short, NO from eNOS possesses the following effects: inhibition of platelet aggregation, vascular wall adhesion [98-100] and leucocyte adhesion to the vessel wall which are early events in the development of atherosclerosis; representing a critical factor for adaptive vascular remodeling to chronicle changes in blood flow [101]; controlling expression of genes involved in atherogenesis and angiogenesis post-ischemia [102]. The abrupt reduction on the bioavailability of eNOS-derived NO is observed after experimental myocardial infarction and in humans under heart failure condition [103, 104], contributing to impaired neovascularization [105]. Accordingly, endothelial NO can reduce the chances of angina episodes, once their effects are also correlated to ischemic-related angina.
Overall, nNOS and eNOS may have distinct roles in the physiological regulation of human microvascular tone in vivo [106]. Interestingly, low levels of nNOS have been shown in vascular smooth muscle cells as responsible to preserve some degree of vasodilatation when the predominant eNOS becomes dysfunctional [55, 107].
Hypertensive patients with metabolic syndrome and also patients with vascular diseases such as atherosclerosis show endothelial dysfunction due to reduced NO bioavailability and consequently impaired endothelium-dependent vasodilatation [108] associated with increased ROS production. There are several enzymatic systems potentially producing ROS in the vessels, including the Nicotinamide Adenine Dinucleotide Phosphate Hydrogen (NADPH) oxidases, XO mitochondrial respiratory chain, uncoupled eNOS [109] and nNOS [110-116]. Of these, NADPH oxidases are considered primary importance for ROS generation. Several isoforms of O2−•-producing NADPH oxidase are expressed in endothelial and smooth muscle cells, as well as in the adventitia layer [55].
The eNOS and nNOS produce large amounts of ROS when deprived of their critical cofactor BH4 or their substrate L-arginine [110-116]. An important stage of the electron transfer occurs in the Heme domain, which receives electron and enables oxygen binding. The reduction directly to the Heme domain is faster than Flavin reduction through the BH4 cofactor. Despite this faster process, the catalytic cycle can only proceed if BH4 was reduced. This difference leads to limitation of the reduced oxygen species productions by heme reduction. In hypertensive vessels Fig. (2), the disruption in electron flowing rather results in reduction of O2 at the prosthetic heme site than formation of NO [117]. The importance of the O2−• formation is due to the BH4 oxidation. Some in vivo studies [118] provided a mechanism for the predisposition to atherosclerosis suggesting NADPH oxidase as the initial source of ROS leading to BH4 oxidation. Endothelial and vascular smooth muscle cells-derived NADPH oxidase produces superoxide, respectively in early and advanced atherosclerosis stages [119]. Despite this knowledge, Laursen et al. [120] described ONOO− as more potent BH4 oxidant than O2−• in hypertension. Indeed, myriad toxic effects of NO are recognized due to the subsequent generation of ONOO− [121, 122] involved in inflammatory conditions [123, 124], neurodegenerative diseases [125, 126] and cardiovascular diseases [118, 120]. Therefore, NADPH oxidase- O2−• may not be an oxidant as relevant in hypertension as ONOO−.
Similarly, the Cyt-C oxidase is a functionally competent ONOO− reductase. It is suggested an enhanced NO production through a positive feedback mechanism for NO2−-derived mitochondrial NO on a Cyt-C oxidase subunit. This protein recruitment is in state-dependent hypoxia; therefore, Cyt-C functional role is in hypoxic signaling events [127].
CONCLUSION
Manifold studies have proved or suggested the control and influences on blood pressure by NOS isoenzymes and have made correlation between NOS-NO and ischemic angina. This is because both, hypertension and ischemic angina, are part of a major MS that affect not just NO production by NOS, but also enzymes pathways, such Cyt-C, important to strictly anoxic conditions [16, 18, 128]. Due to this, it is still important to generate correlations between the many enzymes pathways already described on literature and angina for a better and more complete understanding of CVD.
In the vascular endothelium, BH4 mediates coupling of O2 reduction to heme-catalyzed L-arginine oxidation to form NO and L-citrulline [50]. In patients with MS, there is an inherently systemic inflammation and high risk of CHD [50]. The usual result is atherosclerosis in the coronary arteries leading to NADPH oxidase functioning and ROS products. Beyond ROS strengthen vascular lesions and NADPH oxidase functions, O2−• and ONOO− oxide BH4. Even though the pathophysiologic control of endothelial BH4 levels in humans is poorly known, assembling the information described in the literature databases turns possible to have a better insight about the NO roles in cardiovascular symptoms such as angina.
LIST OF ABBREVIATIONS
| BH4 | = Tetrahydrobiopterin |
| cGMP | = Cyclic guanosine monophosphate |
| CHD | = Coronary heart disease |
| CNS | = Central nervous system |
| Cyt-C | = Cytochrome C |
| eNOS | = Endothelial - Nitric Oxide Synthase |
| GC | = Guanylyl cyclase |
| iNOS | = Inducible - Nitric Oxide Synthase |
| MB | = Methylene Blue |
| mRNA | = Messenger RNA |
| MS | = Metabolic Syndrome |
| mtNOS | = Mitochondrial - Nitric Oxide Synthase |
| NADPH | = Nicotinamide Adenine Dinucleotide Phosphate Hydrogen |
| NCBI | = National Center for Biotechnology Information |
| nNOS | = Neuronal - Nitric Oxide Synthase |
| NO | = Nitric Oxide |
| NO2− | = Inorganic nitrite |
| NO3− | = Nitrate |
| NOS | = Nitric Oxide Synthase |
| O2 | = Molecular oxygen |
| O2−• | = Superoxide anion |
| ONOO− | = Peroxynitrite |
| ROS | = Reactive Oxygen Species |
| XO | = Xanthine oxidase |
| XOR | = Xanthine oxidoreductase |
AUTHORS’ CONTRIBUTIONS
Leticia Gonçalves and Carolina Baraldi Araujo Restini have made substantial contributions to conception and design and interpretation of material collected, as well as in drafting the manuscript, revising it critically for important intellectual content and have given final approval of the version to be published. Therefore, they have agreed to be accountable for all aspects of the work and ensuring questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
CONFLICT OF INTEREST
The authors declare that they have no significant competing financial, professional or personal influencing the performance or presentation of their work described in this manuscript.
ACKNOWLEDGEMENTS
The authors would like to thank to Dr. Reinaldo B. Bestetti and Dr. Suzelei de Castro França by the motivation to study the present aspects of NO pathways.

