All published articles of this journal are available on ScienceDirect.

The Endothelium and Its Role in Regulating Vascular Tone
Authors Info & Affiliations
Abstract
The endothelium forms an important part of the vasculature and is involved in promoting an atheroprotective environment via the complementary actions of endothelial cell-derived vasoactive factors. Disruption of vascular homeostasis can lead to the development of endothelial dysfunction which in turn contributes to the early and late stages of atherosclerosis. In recent years an increasing number of non-invasive vascular tests have been developed to assess vascular structure and function in different clinical populations. The present review aims to provide an insight into the anatomy of the vasculature as well as the underlying endothelial cell physiology. In addition, an in-depth overview of the current methods used to assess vascular function and structure is provided as well as their link to certain clinical populations.
THE ENDOTHELIUM
Once considered as a simple barrier between the blood and vessel wall, the endothelium is now regarded as a dynamic organ which lines the entire vascular system [1]. Endothelial cells are located on the intima – which is the inner lining of the vasculature and they control vascular function by responding to various hormones, neurotransmitters and vasoactive factors which affect vasomotion, thrombosis, platelet aggregation and inflammation [1]. The balanced production of these vasoactive factors is atheroprotective, whereas a damaged endothelium causes disrupted production of these factors. The ensuing imbalance leads to endothelial dysfunction (ED), which is an early indicator of atherosclerosis [2]. Endothelial cells are located on the intima of all vessels (described in detail below), but display different structures and phenotypes depending on vessel type [3]. Endothelial cells in arteries and veins appear more continuous and thicker than those in capillaries which are fenestrated and thinner to allow for exchange of metabolites and gases [4]. In addition, endothelial cells can display heterogeneous responses to stimulation in different vascular beds, and even in different sections of the same vascular bed [5-7]. This suggests that ED may occur in selective vascular beds too [7].
ANATOMY AND PHYSIOLOGY OF THE BLOOD VESSELS
The blood vessels provide the main link between the heart and the tissues. The vascular wall is made up of three layers; the intima (inner layer), the tunica media (middle layer) and the tunica externa (outer layer) [8]. The blood vessels are divided depending on function, location and size into arteries, capillaries and veins.
VASCULAR ANATOMY
The main function of the arteries is to supply the organs with blood. Given the high pulse pressure in the arteries their walls are thicker than in other vessels. Arteries can be divided into conducting arteries, conduit arteries and resistance arteries based on their position in the arterial tree. Conducting arteries are the largest arteries in the body and have a large amount of elastic tissue which allows the vessel to expand and recoil to dampen out the oscillatory changes in blood pressure as a result of intermittent ventricular contractions. Examples of conducting arteries include the aorta, pulmonary artery and carotid artery [9]. Conducting arteries branch into conduit arteries such as the brachial, radial and femoral arteries, and the function of these arteries is to direct blood to specific regions of the body [10]. The conduit arteries further divide into the resistance arteries which are responsible for adequately perfusing the organ tissue with blood and form part of the microcirculation. They consist mainly of smooth muscle cells which are highly innervated by sympathetic nerves, allowing the arterioles to regulate bloodflow to the tissue by dilating or constricting in response to sympathetic (de)activation [4]. Another stimulus that can cause dilation of arterioles is shear stress (the dragging frictional force exerted on the vessel wall by laminar blood flow) [11]. The site of tissue perfusion occurs in the capillaries, which like arterioles are part of the microcirculation [4]. The main function of the capillaries is to enhance the diffusion of gases, metabolites and nutrients between the blood and the tissue. This is achieved by capillary walls which consist of a single layer of endothelial cells, thus, shortening the diffusion pathway between the blood and tissue fluid. Efficiency of diffusion is further enhanced by the slow bloodflow which helps to increase the time available for diffusion [12]. Once gaseous exchange occurs, the blood containing metabolites flows into venules – where further gaseous exchange may also take place. The venules feed into the peripheral veins and then into the superior and inferior vena cavae, which are connected to the heart. In general, the diameter of veins increases with increased proximity to the heart. Given the lower blood pressure in the venous system compared to the arterial system, the vessel walls of veins are thinner and more compliant than arterial walls. This means that veins can accommodate large volumes of blood with only small increases in pressure. Mechanisms such as the skeletal muscle pump and respiratory pump as well as sympathetic nervous activation enable veins to return blood back to the heart. In addition, veins contain valves to prevent backflow of blood while smooth muscle cells in the vascular wall allow veins to constrict and increase the blood pressure, both of which increase venous return [13].
REGULATION OF VASCULAR TONE
The endothelium releases various vasoactive factors. These can be vasodilatory factors such as nitric oxide (NO), prostacyclin (PGI2) and endothelium derived hyperpolarizing factor (EDHF) or vasoconstrictive factors such as thromboxane (TXA2) and endothelin-1 (ET-1). These factors are discussed in greater detail below.
a). Nitric Oxide
Nitric oxide (NO) is an endothelium-dependent vasodilator of the underlying smooth muscle and was first identified by Furchgott and Zawadzki [14]. NO has been shown to play an important role in the maintenance of basal vasodilator tone of the blood vessels [15]. NO is formed under the influence of the enzyme nitric oxide synthase (NOS), which converts the amino acid L-arginine to NO [16]. Three isoforms of NOS exist: neuronal isoform (nNOS) which produces NO to act as a neuronal messenger that regulates synaptic neurotransmitter release [17], macrophage or inducible isoform (iNOS) which is only expressed in cells that have been exposed to inflammatory mediators or other injurious stimuli that activate the macrophages [18], and endothelial NOS (eNOS) which produces nitric oxide in the vasculature [19]. The isoforms are classified by the cells they were originally found in, although, it is now known that expression of these isoforms also occurs in other cells, such as cardiac myocytes [20], skeletal muscle, blood platelets and the hippocampus [21]. Considering that the ability of a blood vessel to dilate is largely dependent upon the activity of eNOS, the present discussion will focus on this isoform.
Inactive eNOS is bound to the protein caveolin and is located in small invaginations in the cell membrane called caveolae [22]. When intracellular levels of Ca2+ increase, eNOS detaches from caveolin and is activated [22]. NO agonists can influence the detachment of eNOS from caveolin by releasing Ca2+ from the endoplasmic reticulum (Fig. 1) [23]. Examples of such NO agonists include bradykinin (BK), acetylcholine (ACh), adenosine tri-phosphate (ATP), adenosine di-phosphate (ADP), substance P and thrombin [24]. Once intracellular Ca2+ stores are depleted a signal (thus far unidentified) is sent to the membrane receptors to open Ca2+ channels allowing extracellular Ca2+ into the cell [25, 26]. This process of Ca2+ regulation is known as store-operated Ca2+ entry or capacitative Ca2+ entry [27]. Ca2+ attaches to the protein calmodulin in the cytoplasm of the cell, after which it undergoes structural changes which allows it to bind to eNOS [28]. eNOS then converts L-arginine into NO [16]. This pathway of NO production is represented in Fig. (1) below. It is important to highlight that this mechanism of NO production is dependent on the levels of intracellular Ca2+ in the endoplasmic reticulum as well as Ca2+ which diffuses into the cell from extracellular stores. A reduction in Ca2+ causes the calcium-calmodulin complex to dissociate from eNOS, which in turn binds with caveolin and becomes inactivated [28].
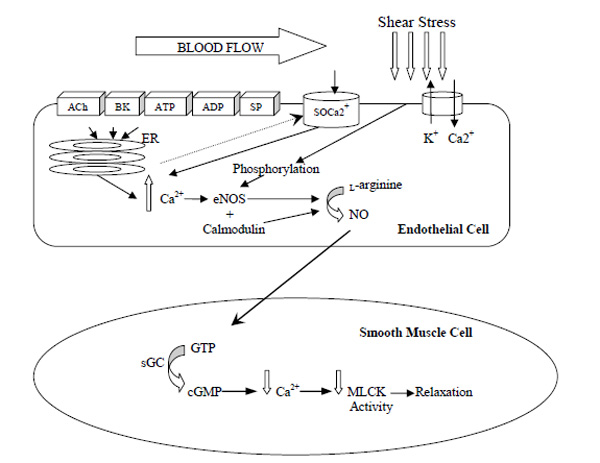
Endothelial nitric oxide production and it actions in the vascular smooth muscle cell. ACh= acetylcholine; BK= bradykinin; ATP= adenosine triphosphate; ADP= adenosine diphosphate; SP= substance P; SOCa2+= store-operated Ca2+ channel; ER= endoplasmic reticulum; NO= nitric oxide; sGC= soluble guanylyl cyclase; cGMP= cyclic guanosine-3’, 5-monophosphate; MLCK= myosin light chain kinase. *When Ca2+ stores of the endoplasmic reticulum are depleted a signal is sent to SOCa2+ channel which allows extracellular Ca2+ into the endothelial cell.
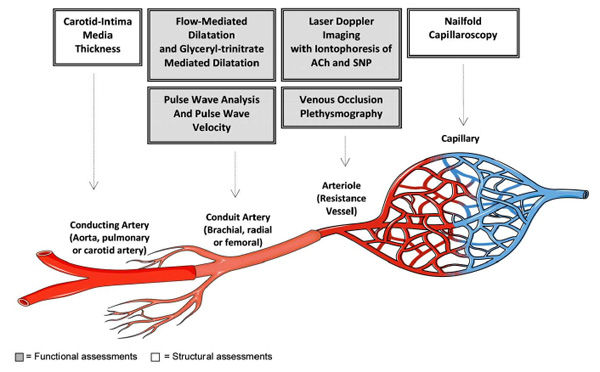
An overview of the assessments for endothelial function and vascular structure performed in different vascular beds. ACh = Acetylcholine, SNP = Sodium nitroprusside.
The short term increase in NO is dependent on the intracellular Ca2+ but once this decreases additional mechanisms are activated to regulate NO production. One such mechanism is the phosphorylation of eNOS [29]. Phosphorylation of eNOS occurs via protein kinases [18], such as protein kinase A [23] and cyclic guanosine-3’, 5-monophosphate (cGMP) protein kinase dependent II [29]. Shear stress initiates eNOS phosphorylation by the actions of protein kinase B (Akt) [30].
Shear stress results from increased bloodflow in the vessel and can increase NO production by eNOS phosphorylation but also through stimulating endothelial cell receptors by allowing the transfer of blood-borne agonists to attach to endothelial cell receptors and increase intracellular Ca2+ [31]. In particular, shear stress activates specialised Ca2+-activated K+ channels on the endothelial cell surface, causing K+ efflux and Ca2+ influx into the cell [32] (Fig. 1). The contribution of Ca2+ and eNOS phosphorylation to NO production is dependent on the duration of the shear stress. For example, intracellular Ca2+ release is dependent on shear stress of short durations [33], whereas shear stress of longer durations (>30 minutes) can deplete intracellular Ca2+ stores, and so NO production is dependent on eNOS phosphorylation [34].
Once synthesized, NO diffuses across the endothelial cell into the adjacent smooth muscle (Fig. 1), where it binds to the enzyme soluble guanylyl cyclase (sGC) [35]. The now activated enzyme increases the conversion rate of guanosine triphosphate (GTP) to cGMP, which decreases smooth muscle tension [36]. Further, cGMP reduces Ca2+ release from the sarcoplasmic reticulum in the smooth muscle cell [37], and also helps to restore Ca2+ to the sarcoplasmic reticulum [38]. Both actions reduce the contraction of smooth muscle cells.
The mechanisms described above are continuously active and produce NO to maintain basal vasodilator tone. By inhibiting NO activity using NG monomethyl-L-arginine (L-NMMA), a dose dependent increase in blood pressure was found due to the vessels constricting, which was reversed when NO was administered [39]. These findings highlight the importance of NO in maintaining resting vasodilator tone. However, the vessel is also capable of dilating in the absence of NO. After removal of or damage to the endothelium, administration of glyceryl trinitrate (GTN) can still result in vasodilatation [15].
The mechanism by which GTN causes vasodilatation is not clear. Several researchers have suggested that GTN undergoes bioconversion to NO [40-42], but not all agree, as GTN has been found to cause vasodilatation without increasing NO [43]. Further, the breakdown products of GTN have been shown to activate sGC [44]. It is worth noting that other vasoactive agents such as calcium ionophore A23187 and isosorbide-dinitrate induce vasorelaxation without an increase in NO concentration [24]. Therefore, NO does not seem to be the only agent that can activate the sGC-cGMP pathway. Further research is needed to identify the precise mechanism of the agents, in particular, more research is needed in vivo due to the differences in response between intact or a denuded endothelium [1].
Aside from vasodilatation, NO is also involved in preventing platelet and leukocyte activation and adhesion to the vessel wall [45, 46]. When the endothelium is damaged, the subsequent inflammation causes an increase in leucocytes at the damaged site [47]. Inflammatory mediators such as TNF-α, interleukin-1 (IL-1) and chemokines stimulate the release of iNOS [48], which prevents leucocytes from adhering to the endothelium and reduces inflammatory mediators [49], as well as down-regulating and reducing the expression of adhesion molecules [50].
b). Prostacyclin and Thromboxane A2
The synergistic actions of two prostanoids, prostacyclin (PGI2) and thromboxane (TXA2) also regulate vascular function [51]. Their production is catalysed by cyclooxygenase (COX) enzymes, of which there are two isoforms COX-1 and COX-2 [52]. COX-1 is expressed continuously in endothelial cells, whereas COX-2 is only expressed when the endothelium is damaged and exposed to inflammatory cytokines [53, 54].
COX-2 converts arachidonic acid to prostaglandin H2 (PGH2), which is then synthesised into PGI2 by prostacyclin synthase [55]. PGI2 binds to the prostacyclin receptors (IP) [56], which are located on both platelets and vascular smooth muscle cells [57]. Activation of platelet IP receptors leads to inhibition of platelet aggregation [58]. PGI2 binding to the smooth muscle cell IP receptor activates adenylate cyclase which induces the synthesis of cyclic adenosine monophosphate (cAMP) [59]. cAMP then activates protein kinase A, which allows relaxation of the smooth muscle in the same way as it does for NO [60, 61]. It is worth noting that in the presence of NO, blocking PGI2 production has no effect on vasodilatation [62]. However, when NO is blocked, the residual dilation is due to increased PGI2 synthesis [63], suggesting that PGI2 plays a compensatory role in dilation of the vessel when NO is reduced.
In contrast to PGI2, TxA2 causes platelet aggregation and vasoconstriction [64]. COX-1 converts arachidonic acid to PGH2, after which TxA2 is synthesised by thromboxane synthase [51]. TxA2 mediates its effects by its actions on thromboxane-prostanoid (TP) receptors which are located on platelets and their activation causes platelet aggregation [53]. The TP receptor is also found on smooth muscle cells and is involved in increasing intracellular Ca2+ levels in the smooth muscle, leading to vasoconstriction [65].
The balance in the activity of PGI2 and TxA2 helps to maintain homeostasis in the healthy vessel. The importance of this balance becomes evident when using selective COX-2 inhibitors to reduce inflammation, which decreases the production of PGI2 without affecting the production of TXA2 [66]. Thus, by administrating COX-2 inhibitors, TXA2 will cause vasoconstriction and platelet aggregation which is unopposed by PGI2, increasing the risk for cardiac events [67].
c). Endothelin-1
Endothelin (ET) is a vasoconstrictor which is expressed in the body in three isoforms, ET-1, ET-2, and ET-3 [68]. Endothelial cells only release ET-1, thus the present discussion will focus only on this isoform. ET-1 is produced by converting Big ET-1 to ET-1 by endothelin converting enzyme [69]. Regulation of ET-1 production as well as its release is stimulated by inflammatory cells such as interleukins and TNF-α and decreased by NO and PGI2 [68]. Shear stress causes a decrease in ET-1 expression, after initially promoting it. ET-1 receptors have been identified both on smooth muscle cells (ETA and ET-B2) and endothelial cells (ET-B1) [70, 71]. The distribution of the different ET-1 receptors is dependent on the type of vascular bed, as veins show a reduced ETA:ETB receptor ratio compared with arteries [72]. When ET-1 binds to ETA or ET-B2 receptors, smooth muscle Ca2+ channels open allowing extracellular Ca2+ into the cell. This causes vasoconstriction in a similar way as TxA2. Activation of ET-B1 receptors on the endothelium causes vasodilatation by inducing the release of NO and PGI2 [73, 74]. In ED, ET-B1 receptors on the endothelial cells are downregulated, while ET-B2 receptors on smooth muscle cells are upregulated, thus enhancing vasoconstriction [75, 76].
The effect of each receptor on the vasculature has been explored in patients with heart disease and in healthy participants. Selectively blocking ETA receptors in participants with ED reliably leads to vasodilatation [76]. However, blocking both ETA and ETB receptors in participants with ED results in greater vasodilatation than blocking ETA receptors only [76]. This finding suggests that the upregulation of smooth muscle ETB receptors has an additive effect on vasoconstriction in individuals with ED [75, 76]. In healthy participants blocking ETB receptors leads to vasoconstriction [77], therefore, ETB receptors located on the endothelium predominantly regulate endothelial function in this group.
Apart from its vasoactive effects, ET-1 also causes inflammation and smooth muscle cell proliferation in the vessel. Binding of ET-1 to ETA receptors activates macrophages, increases neutrophil-vessel wall interactions, and elevates free radical concentrations, all of which lead to ED [78]. ET-1 causes smooth muscle cell proliferation by binding to ET receptors [79] or activating other growth factors such as platelet-derived growth factor [80]. This results in an increase in the intima-media thickness of the vessel wall [81], which can be reduced by blocking ET-1 receptors [82]. In addition, inhibition of ETA receptors in diseased vessels can reduce atherosclerosis, which again suggests that ETA receptors are active during ED [83].
d). Endothelium-Derived Hyperpolarising Factor
Endothelium-derived hyperpolarising factor (EDHF) is a yet unidentified vasodilator substance which hyperpolarises the underlying smooth muscle by making the membrane potential of the cell more negative [84]. EDHF is released when endothelial cells are activated by agonists such as BK and ACh [85]. NO and PGI2 can also dilate the vessel by hyperpolarising the smooth muscle cells, albeit for a short period before the mechanisms discussed above take over [86]. However, when NO and PGI2 are inhibited hyperpolarisation still occurs, suggesting the involvement of a third hyperpolarising factor [87]. A number of pathways have been implicated in causing the hyperpolarisation. Although the exact pathway is still unknown, attention so far has been paid to three factors in particular.
Activation of endothelial receptors and the subsequent increase in Ca2+ levels causes K+ efflux from the cell [88]. The smooth muscle cell responds to changes in the extracellular K+ levels and also releases K+ out of the smooth muscle cell causing hyperpolarisation [89]. The change in the membrane potential of the smooth muscle cell reduces intracellular Ca2+ levels, resulting in relaxation [88].
Epoxyeicosatrienoic acids (EET) are products of arachidonic acid metabolism [90]. Although synthesised in the endothelial cell, they act by increasing K+ efflux from the smooth muscle cells resulting in hyperpolarisation and relaxation [91, 92]. However, in vessels where EET activity is inhibited, hyperpolarisation still occurs [93], suggesting that other mechanisms must be involved in hyperpolarising the smooth muscle cells.
Gap junctions are intercellular channels which can transfer signals from the endothelial cells to the smooth muscle cells [94]. In particular, gap junctions may transfer K+ ions from the smooth muscle cells into the endothelial cell [95]. However, since most studies have only transferred artificial dye between the two cells it is difficult to establish exactly what is transferred under normal conditions.
TECHNIQUES TO ASSESS ENDOTHELIAL FUNCTION
Endothelial function is most commonly assessed in the peripheral circulation as direct assessment of endothelial function in the coronary arteries is highly invasive and associated with considerable risk for the participant. Several studies have reported close correlations between peripheral and coronary endothelial function [96-98]. In addition, assessments of endothelial function are good predictors of future cardiac events in individuals at risk of CVD and those with established CVD [99, 100], and ED is common in individuals with CVD risk factors [101]. Most assessments of endothelial function involve the measurement of dilation in response to a stimulus, with impaired vasodilatation indicative of poor endothelial function. However, impaired vasodilatation can be the result of either the endothelium not sending the signals to the smooth muscle or of the smooth muscle cells not being able to respond to the signal and dilate. Therefore, in order to distinguish between ED and smooth muscle dysfunction, endothelium-dependent and endothelium-independent vasodilatation are typically assessed. Techniques that assess endothelial function in different vascular beds is shown in Fig. (2) and described in more detail below.
ASSESSMENT OF MICROVASCULAR ENDOTHELIAL FUNCTION
a). Iontophoresis
The assessment of NO bioavailability in the microvasculature is conducted using iontophoresis [102]. Iontophoresis uses a small electrical current to pass negatively and positively charged vasoactive agents through the skin into the resistance vessels on the basis that like charges repel each other [103]. The amount of the agent that is delivered to the vessel depends on the density and duration of the current. The two most common agents used to test endothelial function are ACh and SNP [104]. The assessment is usually carried out in the forearm. Laser Doppler techniques are used to assess the perfusion in response to iontophoresis. Laser Doppler flowmetry (LDF) assesses perfusion of the vessel over a single point on the forearm [105]. Perfusion can also be assessed by Laser Doppler imaging (LDI) which uses the same principles as LDF, but rather than scanning one point, a whole area of the forearm can be assessed [106].
The ACh and SNP are administered in small chambers which are attached to the volar aspect of the forearm by watertight adhesive pads. The anodal chamber contains ACh, while SNP is present in the cathodal chamber. Both chambers are connected to an iontophoresis controller which delivers the current [105]. The vasoactive agents can be dissolved in fluid known as vehicles, e.g. deionised water or saline. However, these vehicles can also increase skin perfusion [107]. It has been suggested that use of a lower current density reduces the vasodilatory effects of the vehicles, but drug administration is also reduced [108]. However, a higher current density can be used with 0.5% sodium chloride (NaCl), as it does not elicit a vasodilatory response at this concentration [107]. External factors such as time of day, and menstrual cycle can affect microvascular bloodflow [109, 110]. Therefore, it is advisable to follow established guidelines when administering this test [111].
b). Forearm Blood Flow and Venous Occlusion Plethysmography
Endothelial function of the forearm resistance vessels can be assessed using venous occlusion plethysmography (VOP) [112]. This assessment stops venous return from the forearm, while allowing arterial inflow; blood can enter the forearm but cannot escape resulting in a linear increase in forearm volume with time which is proportional to the incoming arterial blood flow [112]. The halt in venous return is achieved by inflating a blood pressure cuff placed around the forearm to below the diastolic blood pressure (typically 40mmHg) for 10 seconds, followed by 5 seconds of cuff deflation. The hand is excluded from the measurement by inflating a blood pressure cuff which is placed around the wrist to suprasystolic pressures. This reduces the variation in blood volume due to a high proportion of skin blood vessels susceptible to temperature variations. VOP can be assessed using automated equipment which can precisely control the time for cuff inflation and deflation. The increase in forearm volume is assessed by mercury in rubber strain-gauge plethysmograph placed around the widest part of forearm. An increase in the length of the strain-gauge is detected by a change in electrical resistance and represents an increase in forearm blood flow (FBF). It is also important to assess FBF in the contra-lateral arm so that time-dependent changes in basal blood flow due to arterial pressure fluctuations can be accounted for [112]. The FBF response can also be assessed in response to Intra-brachial infusion of various vasoactive agonists (ACh, substance P, bradykinin) or antagonists (L-NMMA, indomethacin) [113].
c). Nailfold Capillaroscopy
Nailfold capillaroscopy is a technique to assess capillary structure [114]. The technique involves the application of immersion oil to the nailfold epidermis of all ten fingers. The nailfold is then placed under a microscope and abnormalities in the capillaries are characterised according to their size, number and structural characteristics. Capillaroscopic abnormalities can be classified into three stages (early, active and late). The earliest change to capillary structure is an enlargement in their size. A reduction in capillary number and structural impairments are seen in the active and later stages of microangiopathy [114].
ASSESSMENT OF MACROVASCULAR ENDOTHELIAL FUNCTION
a). Flow-Mediated Dilatation
Flow-mediated dilatation (FMD) is a technique that increases blood flow through an artery to cause vasodilatation on the principal that the increased bloodflow produces shear forces on the endothelium and subsequently stimulates endothelial cells to release NO [30]. As indicated previously, reduced vasodilatation following an increase in shear forces is representative of impaired NO bioavailability [115]. Therefore, FMD is a good surrogate marker of NO bioavailability. The FMD protocol involves a 2 minute baseline ultrasound scan of the brachial artery, after which a cuff placed around the wrist is inflated to 300 mmHg for 5 minutes. This causes tissue ischemia and dilation of downstream resistance vessels via auto-regulatory mechanisms. When the cuff is released a sudden increase in bloodflow (reactive hyperaemia) through the brachial artery fills the dilated resistance vessels and in doing so exerts shear stress on the endothelial cells [116]. The resulting dilation, which peaks at 60-90 seconds after cuff release is dependent on NO activity [117]. FMD is expressed as the maximum percentage change in vessel diameter after cuff release relative to baseline vessel diameter [118], with a low percentage indicating poor endothelial function [113]. FMD is typically carried out in the brachial artery using high resolution ultrasound to assess the vessel diameter, but other arteries such as the radial and femoral artery have also been used to measure FMD [117]. Another method to quantify the dilation is strain-gauge plethysmography, with the strain-gauge detecting the change in arm circumference following an increase in blood flow [119].
The protocol used for FMD is important as both occlusion duration and cuff placement have been shown to influence FMD. Five minutes of limb occlusion is adequate to evoke endothelium-dependent dilatation, with longer cuff durations showing a non-NO response [120]. Similarly, the placement of the cuff around the wrist is dependent on NO, whereas cuff placement on the upper arm is only partially mediated by NO [121]. Further, FMD responses can be affected by external factors such as sleep deprivation [122], hyperhomocysteinemia [123], caffeine [124], smoking [125], antioxidant therapy [126], menstrual cycle [127] and time of day [128]. Accordingly, it is important to control these factors [116].
b). Glyceryl Trinitrate
As described earlier, GTN produces dilation of the vessel by acting directly on the smooth muscle cells [15]. As such, the vasodilatory response to activated smooth muscle cells can be assessed by GTN administration. GTN is commonly administered as a vasodilator to cardiac patients presenting with angina as a tablet or oral spray, both of which are placed or sprayed directly under the tongue. Typically, the assessment is carried out for 3-4 minutes, which is the time necessary for the vessels to reach peak dilatation [129].
c). Arterial Stiffness
Each time the heart contracts pressure waves are sent throughout the vasculature and the compliant arterial wall serves to dampen pressure oscillations that stem from the aortic root to aid smooth delivery of bloodflow to the tissues [9]. When the pressure waves reach branch points in the vasculature they are reflected back towards the heart. In a healthy individual the wave arrives during diastole to aid filling of the coronary vessels. However, in individuals with reduced arterial elasticity the pressure wave returns to the heart much quicker and arrives during the systolic phase of the cardiac cycle. This serves to augment the afterload (the pressure the heart has to overcome to open the aortic semilunar valve) [8]. Some notable complications of arterial stiffness include insufficient myocardial perfusion leading to angina or a myocardial infarction, and left ventricular hypertrophy which may result in heart failure [130]. It is therefore not surprising that assessments of arterial stiffness are associated with a number of CVD risk factors such as ageing, smoking, hypertension and dyslipidaemia [130]. Stiffening of the vascular wall can occur due to a reduction in NO production from endothelial cells, loss of smooth muscle tone [131], as well as degeneration of elastin fibres and increased collagen deposition in the vascular wall [132]. Consequently, arterial stiffness is dependent on functional and structural changes in the vasculature.
A number of techniques can be used to assess arterial stiffness non-invasively from the peripheral circulation. The most widely used techniques at present are pulse wave analysis (PWA) and pulse wave velocity (PWV) due to their good reproducibility and ease of use [133]. These assessments have been reported to associate with coronary microvascular endothelial function [134]. PWA is the single measurement of radial artery pressure waveforms which are recorded using a transducer which flattens but not occludes the artery (applanation tonometery). The waveforms are calibrated against the standard brachial blood pressure which gives the maximum (systolic) and minimum (diastolic) points of the pressure curve. The pressure waveform is then mathematically transformed into a central aortic waveform which contains the first and second systolic peaks and displays the augmentation index (AIx). AIx is calculated as the difference between the second and first systolic peaks and is expressed as a percentage of the pulse pressure, with a high value indicating greater arterial stiffness [135]. To obtain PWV readings, arterial pressure waveforms are simultaneously derived from two arteries, usually the carotid and radial arteries, using an applanation tonometer. The distance between the two arteries is then measured and the wave transit time between these two points is recorded to give a quantifiable PWV, with a greater PWV indicating quicker wave reflection back towards the heart and therefore greater arterial stiffness [136].
d). Carotid Intima-Media Thickness
Assessment of carotid-intima media thickness (cIMT) using B-mode ultrasound was first introduced in 1986 by Pignoli and colleagues [137]. The assessment detects thickening of the medial layer of the vascular wall and is a good predictor of cardiac events in patients with early atherosclerosis [138], and is also an important predictor for restenosis in patients who have undergone percutaneous coronary intervention [139]. In addition, increased cIMT has been reported to relate to a number of classical CVD risk factors such as ageing, hypertension, and dyslipidemia [140]. Changes in cIMT represents a sequence of events resulting from a decrease in NO bioavailability as well as an increase in ET-1 levels, which over time increase production of inflammatory cytokines, free radicals, adhesion molecules and thrombotic factors leading to smooth muscle proliferation [141, 142]. Assessment of cIMT is typically performed in the common carotid artery, internal carotid artery and at carotid bifurcation points [143], and each site has a similar ability to predict future cardiovascular events [144].
ENDOTHELIAL DYSFUNCTION IN SELECTED CLINICAL POPULATIONS
a). Endothelial Dysfunction and Cardiovascular Disease
Endothelial dysfunction is evident before the presentation of obstructive atherosclerotic lesions in both conduit and resistance coronary vessels [145], and can even occur in children with a family history of cardiovascular disease [118]. The magnitude of ED increases in line with the accumulation of CVD risk factors in peripheral conduit vessels [146]. Furthermore, endothelial function is a good prognostic marker of future cardiac events in patients with CVD [99]. Administration of L-arginine can increase NO bioavailability and improve endothelial function in patients with CVD risk factors [147]. In addition, medications that control CVD risk factors like anti-hypertensives or statins may also have beneficial effects on endothelial function primarily through decreasing oxidative stress and lipid accumulation [101].
b). Endothelial Dysfunction and Hypertension
In hypertension, the delicate balance between vasodilators and vasoconstrictors produced by the endothelium is disrupted, with disturbance in the NO pathway leading to predominance of vasoconstrictors like ET-1, which contribute to high blood pressure [148]. Even though it is still unclear whether ED is the cause or the consequence of elevated blood pressure , it appears to be an essential factor in hypertension [149]. Studies in humans have reported a significant impairment of the vasodilator response of small resistance vessels to ACh, but not to SNP, in hypertensive patients [150]. Additionally, impaired FMD in the conduit vessels identifies hypertensive patients at increased risk for non-fatal and fatal cardiovascular events [151], whereas the AIx is a predictor of cardiovascular mortality in subjects with essential hypertension [152]. Treatment with angiotensin-converting enzyme (ACE) inhibitors have been shown to improve endothelial function [153]. ACE inhibitors reduce oxidative stress and stimulate bradykinin to help increase NO bioavailability [154].
c). Endothelial Dysfunction and Diabetes
Individuals with type I and type II diabetes have evidence of both microvascular and macrovascular ED [155]. ED can even be evident in healthy individuals with a family history of diabetes [156], suggesting a genetic link. Patients with diabetes often have reduced NO bioavailability which results from increased oxidative stress [157], and oxidation of LDL due to hyperglycaemia [158]. Patients with type 1 diabetes have shown improved endothelial function when taking ACE inhibitors [159], through a reduction in oxidative stress, and an increase in NO bioavailability [154].
d). Endothelial Dysfunction and Inflammatory Diseases
Patients with a variety of inflammatory disorders such as rheumatoid arthritis, bechet’s disease and inflammatory bowel disease are also at an increased risk for developing CVD [160-163]. In RA, the severity of inflammation can impact on the extent of ED [164]. Assessments of carotid atherosclerosis appear to have good prognostic value in patients with inflammatory diseases [165, 166], and as such their use to determine CVD risk has been advocated in such conditions [167]. Use of anti-inflammatory medications can improve endothelial function in the resistance and conduit vessels [168, 169], which further supports the role of inflammation as an important predictor of CVD in inflammatory diseases [170].
SUMMARY
The endothelium is important in maintaining vascular homeostasis and preventing the development of atherosclerosis. However, perturbation of its activity may lead to ED which, if left untreated, could progress to atherosclerotic lesion formation and subsequent cardiac events. Therefore, assessing endothelial function in patients at risk of cardiovascular disease is important to identify vascular abnormalities and may help monitor strategies and interventions that can improve endothelial function and lower CVD risk.

