All published articles of this journal are available on ScienceDirect.

Experimental Models of Duchenne Muscular Dystrophy: Relationship with Cardiovascular Disease
Abstract
Almost every boy that has Duchenne Muscular Dystrophy (DMD) will develop cardiac problems. Whereas, it used to be respiratory problems that was the main cause of death in these DMD boys; with the advent of better respiratory care it is now the cardiac involvement that is becoming the most common cause of their death. Once the heart is affected, there is progressive deterioration in the function of the heart over time. The main problem is the death of the cardiomyocytes. The cause of the cardiomyocyte death is due to the loss of dystrophin, this makes the sarcolemma more susceptible to damage, and leads to a cascade of calcium influx, calcium activated proteases and ultimately the death of the cardiomyocyte. The dead cardiomyocytes are replaced by fibrotic tissue, which results in a dilated cardiomyopathy (DCM) developing, which begins in the base of the left ventricle and progresses to involve the entire left ventricle. The treatments used for the DMD cardiomyopathy are based on ones designed for other forms of cardiac weakness and include ACE-inhibitors and β-blockers. New therapies based around the pathophysiology in DMD are now being introduced. This review will look at the pathophysiology of the cardiac problems in DMD and how the various animal models that are available can be used to design new treatment options for DMD boys.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a lethal X-linked recessive disorder that affects 1 in 3,500 live newborn males [1, 2]. The disorder is caused by mutations in the dystrophin gene, usually deletions or point mutations that lead to a shift in the reading frame and produce a stop codon, thus causing the complete loss or very reduced and deficient synthesis of dystrophin protein, which forms an integral part of the dystrophin-glycoprotein complex (DGC) [3]. A major function of the DGC is to connect the subsarcolemmal cytoskeleton to the extracellular matrix, with dystrophin forming the intracellular link acting like a shock absorber as the cell contracts and moves (Fig. 1). Dystrophin is one of the largest proteins in the body at 427kDa and it is not just expressed in skeletal muscle but also in cardiac muscle as well as in the brain. So although DMD is described as a primary degenerative condition of the skeletal muscle there are additional detrimental effects on the function of the heart and around 90% of DMD boys will develop severe cardiac problems [4]. The female carriers of DMD are usually free of skeletal muscle symptoms, but around 30% will develop some skeletal muscle problems and about 10% of these women will also develop some signs of heart weakness. These manifesting carriers are due to a non-random inactivation of the X chromosomes, leading to a predominant expression of the X chromosome that carries the mutation in the dystrophin gene. Becker muscular dystrophy (BMD) is a milder variant of DMD, also caused by mutations in the dystrophin gene. In BMD the deletions usually do not cause a shift in the reading frame; this tends to produce a functional but truncated version of the dystrophin protein. BMD patients can usually walk and have a near normal life span and there is a 50% chance of them developing cardiac problems [5].
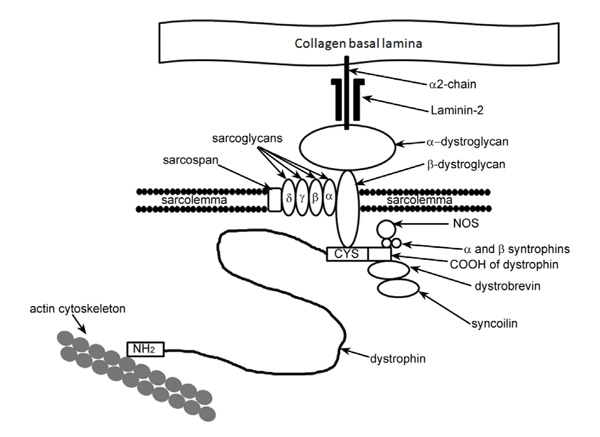
A schematic diagram of the position of dystrophin and its connection with the members of the dystrophin-glycoprotein complex (DGC). Dystrophin acts as a link between the basal lamina and the actin cytoskeleton helping to maintain the integrity of the sarcolemma. The loss of dystrophin compromises the DGC and leads to a more fragile sarcolemma.
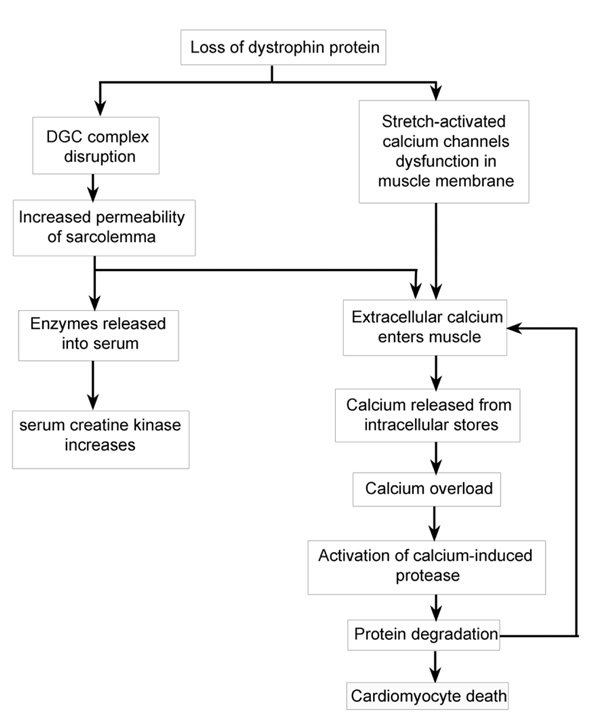
A flow diagram of the known pathways by which the loss of dystrophin or a severely truncated dystrophin leads to the development of cardiomyocyte death.
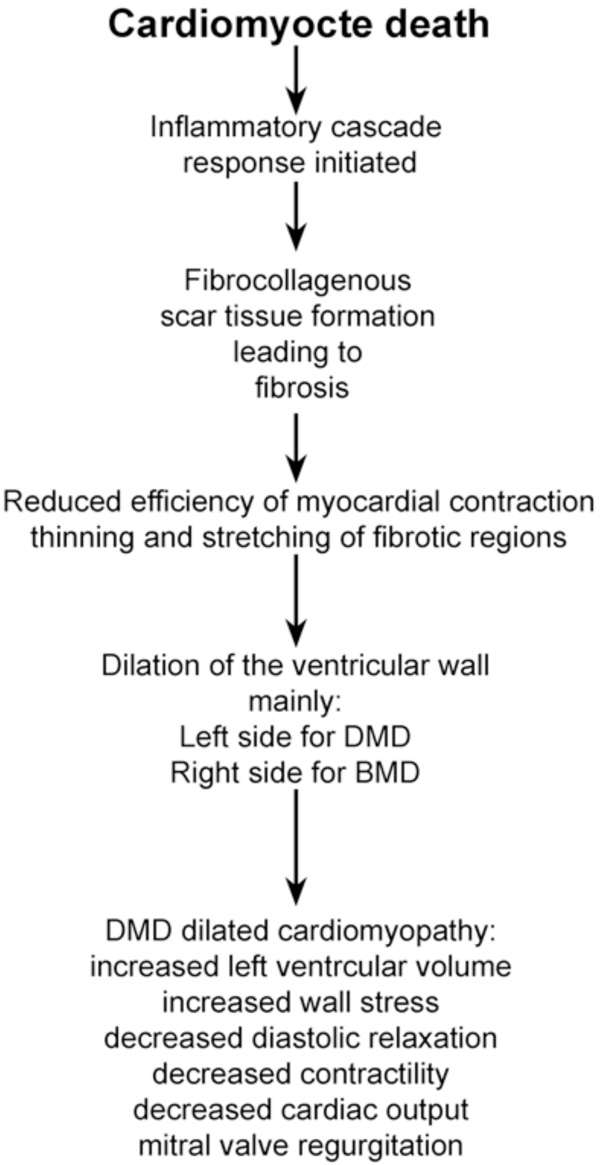
A flow diagram of how the death of the cardiomyocytes caused by the lack of dystrophin then develops into the dilated cardiomyopathy seen in DMD patients.
DMD patients are normally diagnosed clinically between 3 and 6 years of age, presenting with a delay in their motor development and weakness of the skeletal muscle. As the disease progresses the boys have problems walking; this is due to the wasting and atrophy of the muscles and as the muscles become wasted contractures form. The muscle weakness and the contractures together generally result in the boys becoming wheelchair bound around 10-12 years of age. Almost all skeletal muscles are affected in DMD with the muscle groups in the limbs preferentially affected, followed by the trunk muscles, then the respiratory muscles. Patients used to die in their late teens or early twenties mainly from pulmonary problems. However, because of advances in respiratory care, such as nocturnal and continuous 24-hour ventilator support, the life expectancy of DMD patients has increased to their late twenties to mid thirties and some patients may survive into their forties. In these older DMD patients, the major cause of death is now the result of the progression of the cardiac problems [4]. Therefore, the management of the cardiomyopathy is becoming a more important consideration in planning the care of older DMD patients.
Preclinical cardiac involvement has been reported in 25% of patients under the age of 6 years, increasing to 60% of patients between the ages of 6-10 years and then declining in incidence with age [6]. Clinically apparent cardiomyopathy becomes evident after 10 years of age and increases in incidence with age, being present in virtually all patients over 18 years of age [7]. Often the clinical aspects of the cardiac defects associated with DMD are masked by the muscle weakness, which causes reduced physical activity in these boys. The progressive dilated cardiomyopathy (DCM) that develops in DMD boys is presumed to be a secondary consequence of the fibrosis within the myocardium. In both skeletal and cardiac muscle, the function of dystrophin is to protect against contraction-induced damage. In skeletal muscle, there exists a resident stem cell population, the satellite cells, which can repair the damaged fibres even though the repair is still with dystrophin deficient cells. This means that the regenerative capacity of skeletal muscle is severely impaired leading to the eventual replacement of skeletal muscle with fatty and fibrotic material. However, there are no stem cells in the heart to regenerate the damaged cardiomyocytes so damaged cardiomyocytes will tend to die and be replaced by fibrotic tissue earlier than skeletal muscle. Therefore, the heart is potentially at greater risk than the skeletal muscle and the result of this cardiomyocyte damage and replacement with fibrotic material is that currently 10-50% of DMD patients die as a result of heart failure [8, 9].
DYSTROPHIN IN THE CARDIOMYOCYTE
Specifically, the absence of the dystrophin protein in the heart results in the development of a DCM [10]. As with skeletal muscle the loss of dystrophin and the disruption of the DGC (Fig. 1) results in the sarcolemma of the cardiomyocyte becoming more fragile and susceptible to damage from muscle contractions, leading to small tears in the cardiomyocyte membrane. Recent studies suggest that absence of dystrophin may disrupt the function of the sarcolemmal stretch-activated ion channels, which respond to mechanical stress [11]. It appears that the stretch-activated ion channels do not open appropriately in dystrophin deficient cardiomyocytes when they are stretched during ventricular filling; this causes an increase in the influx of calcium into the cardiomyocyte [12]. The tears in the cardiomyocyte cell membrane allow the entry of extracellular calcium into the muscle fibre raising the intracellular calcium levels (Fig. 2). The increase of the intracellular concentration of calcium in the cardiomyocyte leads to the activation of calcium-activated proteases such as calcium-induced calpains, a family of proteases, which will degrade troponin I and thus compromise the contractions of the cardiomyocyte [13-15]. The activation of these calpains also mediate the destruction of the cardiomyocyte plasma membrane proteins, which cause even more holes in the sarcolemma and allowing even more calcium to enter the cardiomyocyte and further damage to the cardiomyocyte. Eventually this chronic calcium overload leads to the death of the cardiomyocyte (Fig. 2) [16]. The increased permeability of the sarcolemma and the degradation of muscle proteins also results in the release of intracellular muscle proteins from the myofibre, such as creatine kinase (CK). Serum CK levels may reach 200-300 times above normal and consistently raised serum CK levels are used as a diagnostic and predictive marker for the extent of the muscle damage in DMD and BMD. This is because the serum CK values vary with the age and the degree of muscle damage and thus correlate with the clinical progression in both DMD and BMD [17].
Cardiomyocytes possess several distinctive characteristics, which contribute to the deterioration of the heart in DMD and BMD patients. It has already been mentioned that there are no stem cells to replace lost cardiomyocytes once they die but in addition there are other unique characteristics that contribute to the development of the DCM in DMD. Firstly, cardiac muscle will repeatedly contract around 86,400 times per day whereas skeletal muscle will only contract intermittently when needed to move a part of the body. These continual contractions result in constant fluxes in the intracellular calcium levels associated with each excitation-contraction cycle, which undoubtedly accelerates the deterioration process within the cardiomyocytes compared to skeletal myofibres. Secondly, the contraction of the cardiomyocytes is based on a calcium-induced calcium release manner, which means that only a small amount of extracellular calcium is needed to enter the cell to induce larger amounts of intracellular calcium to be released into the cytoplasm. When the sarcolemma of the cardiomyocyte becomes leaky because of the lack of dystrophin it means that more extracellular calcium is now able to leak into the cardiomyocyte. The L-type calcium channels can be activated unnecessarily, causing the release of intracellular calcium and actually initiating contraction of the heart, leading to extra contractions of the DMD hearts [18]. These extra contractions caused by the leaky sarcolemma and higher intracellular calcium levels are manifest as arrhythmias, which also directly cause more damage to the cardiomyocytes as more tears will be generated in the membrane. Ultimately, the elevated intracellular calcium concentration activates the cascade of protein degradation and cardiomyocyte death. There is therefore a vicious cycle initiated by the loss of dystrophin plus the cardiomyocyte is particularly susceptible to damage and there is no regenerative capacity in the heart to replace the lost cardiomyocytes all of which leads to the development of the cardiomyopathy.
CARDIOMYOCYTE FIBROSIS AND DILATION OF THE HEART
The cardiomyocyte death usually occurs in very discrete, localized small areas of the heart which have been referred to as ‘microinfarcts’ [19]. These microinfarcts can often be accompanied by chest pain in the patients and diagnosed by the release of cardiac troponin into the serum [19]. Within these focal necrotic areas, there is an inflammatory cascade, which is initiated by the death of the cardiomyocytes. Macrophages migrate into the heart to remove the damaged and dead cells (Fig. 3). Once the macrophages have removed the cardiomyocyte debris, fibroblasts then invade the damaged area and form a fibrocollagenous scar tissue resulting in the deposition of fibrotic tissue in the walls of the heart, which replace the contractile cardiomyocytes. These fibrotic areas are very inflexible compared to the normal cardiac muscle that it replaces and thus there is a reduction in the efficiency of myocardial contraction. For reasons that are still unclear, the fibrosis begins in the left ventricular wall in DMD and in the right ventricular wall in BMD and there is sparing of the atria. In both DMD and BMD, the fibrosis of the heart starts on the outside of the ventricular wall, so starting in the epicardium and progresses into the endocardium [20]. This pattern of fibrosis is unique to the dystrophinopathies [7, 20]. As the fibrosis progressively spreads throughout most of the outer half of the ventricular wall it causes the ventricle to gradually stretch and enlarge, resulting in the ventricular wall becoming thinner, leading to a loss of contractility. The result of this thinning and enlargement of the ventricle is the formation of a DCM in DMD and BMD patients. The volume of the left ventricle will increase because of the enlargement of the chamber but there will be a decrease in systolic function, and there is often mitral valve regurgitation due to the valves no longer meeting because of the enlargement of the ventricle. The result of all these changes is that there is a decrease in the cardiac output and hemodynamic decompensation [21].
DCM PATHOLOGY SPECIFIC TO DMD AND BMD
In addition to the development of the DCM many DMD patients also develop arrhythmias, as a result of the increased intracellular calcium causing extra contractions, and these contribute significantly to the clinical progression. There can be sinus node dysfunction, atrioventricular node dysfunction, atrial fibrillation and ventricular tachycardia/fibrillation, which can develop at any time but which become more common with age and as the damage to the sarcolemma of the cardiomyocytes increases. Screening of all DMD boys is done by echocardiography and electrocardiogram (ECG) and it is recommended that they are screened at regular intervals after the age of 10 and these screens should ideally be performed at least annually [6, 22].
It has been reported that 90% of DMD patients have abnormal ECG readings. From the 12 lead ECG the irregular conduction patterns can be recognized which cause a prominent Q wave in leads I, aVL, V5, and V6, or leads II, III, aVF, V5 and V6 [23]. Tall R waves in V1, and an R/S ratio of greater than 1 [24]. A shortened PR interval has also been reported in about 50% of patients and this is the most common ECG change in DMD patients [23]. Although there do appear to be differences between young and older individuals, with abnormal Q waves or Q/R ratios in younger individuals and a high-grade ventricular ectopy being more frequent in older subjects [25]. Resting sinus tachycardia, a loss of the hearts circadian rhythm, and heart rate variability which is the result of increased sympathetic activity can be observed when Holter ECG monitoring is used in DMD patients [26]. It has also been reported that there is no difference in the ECG findings of DMD patients who have been diagnosed with DCM and those that are still in the subclinical stage, indicating that the ECG changes begin before the progression to the full DCM starts [23]. The use of the ECG findings is therefore not very useful in diagnosing and monitoring the progression of the cardiomyopathy.
In DMD, the myocardial fibrosis and dilation tends to appear first in the left ventricular wall behind the posterior mitral valve leaflet. The fibrosis progresses inferiorly toward the apex of the heart and into the interventricular septum; eventually the entire left ventricle will be affected and the interventricular wall of the right ventricle [7, 27, 28]. In BMD patients, the pattern of the fibrosis progression is similar to that observed in DMD patients but the right ventricle is affected preferentially rather than the left [7]. Echocardiogram and magnetic resonance imaging (MRI) can visualize the posterior thinning of the epicardium, the areas of dyskinesis and akinesis, as well as giving a direct measurement of the systolic and diastolic dysfunction. The E point-to-septal separation (EPSS) is a measure of the minimal separation between the mitral valve anterior leaflet and the ventricular septum during early diastole [29]. An EPSS of greater than 5–5.5 mm suggests that the left ventricle has become more spherical [29]. The sphericity index, calculated by dividing the length of the left ventricle by the width of the left ventricle, is useful in evaluating the shape of the chamber of the left ventricular. A sphericity index of greater than 0.66 suggests DCM [30]. Other signs of dilation include increased left ventricular diameter and volume, decreased shortening and ejection fractions, and development of mitral valve regurgitation.
DIAGNOSIS OF CARDIOMYOPATHY IN DMD PATIENTS
A diagnosis of cardiomyopathy in DMD patients is not easy because they tend to have a reduced exercise tolerance, which masks the cardiac problems. The fact that in DMD patients there is immobility, deformities, and pulmonary failure additionally obscure the clinical and radiographic signs and symptoms of heart failure. The standard diagnostic tool is echocardiography and annual echocardiograms after the age of 10 are recommended. However, echocardiography, although readily available, gives only a crude indication of the heart problems and it is made more difficult in DMD patients by the development of scoliosis, which hampers the accurate measurements of the chambers of the heart.
Multigated cardiac radionuclide ventriculography (MUGA) is not affected by these problems and has been found to be sensitive in detecting subclinical cardiomyopathy in DMD, BMD and female carriers [6, 31]. MUGA was able to detect a predominant systolic and minor diastolic left ventricular dysfunction even in the BMD patients and female carriers where there was no skeletal muscle involvement [31]. However, MUGA is associated with repeated radiation exposure, which becomes more relevant in DMD when annual screening of cardiac function is recommended to monitor the function of the heart and the progression of the cardiomyopathy in DMD patients. Therefore, although MUGA has been shown to be as sensitive as echocardiography at detecting subclinical cardiomyopathy in DMD is has not been proven to be superior to echocardiography in assessing the systolic function. In addition MUGA is not available in all centers and it is expensive to perform.
Tissue Doppler can be used to identify myopathic changes that happen early in the development of the cardiomyopathy and may also reflect the ongoing damage to the heart [27]. MRI has been suggested as another non-invasive method of monitoring the cardiac function in DMD patients. There is no ionizing radiation used in MRI and with the newer MRI machines high-resoultion images can be obtained. The T2 relaxation time can be used to quantitate the tissue characteristics of the heart as it reflects the differences in water and fat content of the muscle [32]. However, MRI is expensive and not all centers have access to such a facility, but it is certainly more sensitive at detecting the cardiomyopathy when it is in the sub-clinical stage than many of the other methods available.
New diagnostic and prognostic biomarker tests using the natriuretic peptides, brain natriuretic peptide (BNP) and N-terminal proBNP (NTproBNP), have proven to be useful diagnostic factors for patients with heart failure [22, 33-35]. In DMD, patients that were not on mechanical ventilation the plasma levels of BNP were associated with systolic dysfunction [36]. The combination of the plasma levels of natriuretic peptides linked to the echocardiogram have proven to be capable of evaluating the left ventricular function to a level that is comparable to the results obtained from MUGA [6, 36]. A further advantage of using the levels of the natriuretic peptides in combination with echocardiograms is that it is less time consuming and does not cause distress to the patients [6]. However, whatever method is employed to test the heart the overriding principal is to decide what degree of weakening of the heart should initiate medical intervention. The traditional view is that treatment should start when there are overt signs of heart weakening and early intervention is inappropriate. This view is now being challenged as more data become available from animal model studies and human patients as to the pathophysiology of the DCM in DMD.
PRESENT TREATMENT FOR DMD CARDIOMYOPATHY
The general therapy for DMD includes the use of glucocorticoids around the age of 5 or 6. The side effects of these drugs limit the effectiveness of long-term use but they have proven to slow the progression in the wasting of the skeletal muscle and can delay the use of a wheelchair by 1 to 2 years. There is now a movement to prolong the use of the steroids even after the boys become wheelchair bound as there is some evidence that it may help to maintain the cardiac and pulmonary function. In addition to the steroids, specific pharmacological treatment should be offered to all DMD and BMD patients with cardiac complications. At present treatment using β adrenergic blockade and inhibition of ACEs have proven to be effective at remodeling the heart [37].
The progressive cardiac fibrosis that develops in DMD eventually leads to increased stress in the ventricular wall, and decreased cardiac output. The decrease in cardiac output leads to activation of the renin-angiotensin aldosterone system (RAAS), which is vital in the regulation of sodium and water in the body. The first RAAS component activated is renin, an enzyme that cleaves angiotensinogen to form angiotensin I. Eventually, the ACE produced by endothelial cells in the lungs, transforms angiotensin I to angiotensin II. Angiotensin II stimulates the adrenal cortex to secrete aldosterone, promoting fluid and sodium retention. Both angiotensin II and aldosterone contribute to the formation of fibrosis and the deposition of connective tissue within the heart. Specifically, angiotensin II acts as a growth factor at sites of tissue repair and enhances the activity of transforming growth factor beta (TGF-β1), which then stimulates the proliferation of the fibroblasts that cause the deposition of the fibrotic material in the walls of the heart [38-40]. Similarly, aldosterone is involved in the synthesis of fibrosis forming collagen [41]. These harmful consequences of RAAS hyperactivity secondary to decreased cardiac output further complicate the myocardial fibrosis resulting from dystrophin deficiency in DMD and BMD patients. Therefore, the use of ACE inhibitors, angiotensin receptor blockers (ARBs), and aldosterone antagonists in DMD and BMD patients with cardiomyopathy is recommended.
An uncontrolled retrospective study showed that echocardiographic parameters, including left ventricular ejection fraction, fractional shortening, and sphericity index, in DMD and BMD patients improved 3 years after administration of either ACE inhibitors alone or the combination of both ACE-inhibitors and β-blockers [42]. Around the same time, a double-blind multicenter study was conducted to evaluate the effect of preventive afterload reduction in muscular dystrophy patients. In this study, DMD patients between 9.5 and 13 years old with normal ventricular function were randomly assigned to receive 3 years of placebo or perindopril, an ACE inhibitor. After this 3-year period, every participant received 2 years of perindopril. At the conclusion of the study, a lower left ventricular ejection fraction was found in those subjects who did not take perindopril for the first 3 years [43]. The findings of this study not only suggested the beneficial effects of perindopril but also encouraged early pharmacological interventions to preserve cardiac function. A later study in 2006 tested another ACE inhibitor, enalapril, and monitored left ventricular systolic dysfunction in DMD patients with DCM, diagnosed based on left ventricular fractional shortening. Normalization of fractional shortening occurred in 43% of the patients on enalapril [44]. This improvement was maintained for up to 4 years and again suggests that earlier intervention may prevent the worsening of the subclinical cardiomyopathy in to full blown clinical DCM in a significant proportion of DMD patients.
New treatments based on the fundamental pathogenesis of DMD are now also being used clinically. As the entry of calcium into the cardiomyocyte is a major feature, leading to the fibrosis of the heart it has been proposed that the use of calcium channel blockers, such as diltiazem, flunarizine, and nifedipine would be beneficial for DMD [45-48]. However, no positive improvements have been reported and so the use of calcium blockers has not proven successful.
The use of low dose potassium sparing diuretics, such as spironolactone or eplerenone has been shown to be able to slow or prevent the development of the fibrosis [49, 50]. The downside of potentially combining spironolactone and ACE-inhibitors is that it can result in the building up potassium in the blood and therefore requires careful monitoring by frequent blood tests. The use of anti-oxidants has also been proposed as useful for both the heart and skeletal muscle pathology in DMD, and as a rule, these have no serious interactions with the other drugs and can be relatively easily introduced into the treatment regime of the DMD boys. However, the downside is that it increases the number of medications that the boys have to take every day.
A major problem in understanding the results of the clinical trials so far tested for treatment of the cardiomyopathy in DMD patients is the still relatively small number of patients that are available to enter trials, and we just do not have the numbers of suitable patients to try all the suggested treatments. In such cases, the testing of potential new treatments in the available animal models of the disease becomes essential. The animal models also allow the pathophysiology of the cardiomyopathy to be investigated further. There are a number of animal models, which are genetic equivalents to the human DMD.
AVAILABLE ANIMAL MODELS OF DMD: ADVANTAGES AND DISADVANTAGES
Cardiomyopathy in the Mdx Mouse
The mdx mouse despite being a genetic homolog to DMD has not been fully validated as a model for dystrophin deficient cardiomyopathy. The relatively mild phenotype and near normal life span of the mdx mouse has prompted the generation of double mutants such as the mdx/utrophin and the mdx/MyoD mice, both of which have a far more severe skeletal and cardiac muscle phenotype to model the development of cardiomyopathy. Both of these double knockouts (DKO) have been proposed as a more accurate model system for the study of the cardiomyopathy that develops in DMD and for the testing of new therapeutic options. However, the mdx mouse does develop cardiomyopathy and can be a suitable model for the human DMD cardiomyopathy although it develops the cardiomyopathy very late in its life [51].
The mdx mouse like the DMD patient has a complete loss of dystrophin from the cardiomyocytes. By 10 months of age the mdx mouse does display signs of a DCM with hypertrophied hearts, which have poor contraction and beat at a slower rate than normal [51]. The contraction speed and the force are significantly altered in the atria even in young mice and this is found before there is any obvious necrosis and resulting fibrosis of the walls of the ventricles of the heart. The fact that there are these early changes, before any major fibrosis, suggests that the impaired function is not a consequence of the fibrosis or cardiomyocyte death but is related to the membrane ion channels that regulate calcium entry into the cardiomyocyte. In the mdx mouse, although the left ventricular systolic function is initially normal in 2-month-old mice, by 1 year old a DCM starts to develop and this is accompanied by interstitial fibrosis [51]. The ECGs of conscious mdx mice younger than 10-12 weeks old are more or less normal but around 10-12 weeks they begin to show similar abnormalities to those observed in DMD patients [52]. Other studies have shown that even at 8 weeks the mdx heart is more susceptible to damage when subjected to mechanical stress [53]. These studies highlight a major consideration when using the mdx mouse for studying the disease processes of DMD - under normal housing conditions the heart and the skeletal muscles of the mdx mouse are not stressed, and there is therefore only minimal damage caused to the heart and skeletal muscles, so the mdx mouse displays a milder phenotype. The reduced stress placed on the heart means that the hearts of the mdx mouse develop cardiomyopathy around 10-12 months of age compared to the relatively earlier development in DMD boys. However, if the heart and the muscles are stressed by making the mdx mouse exercise or by ex vivo manipulations of the heart then the mechanisms for damage are present and the heart and muscles behave more like those of the human DMD patients.
The difference in the speed and pathological location of the cardiomyopathy that develops under normal and exercise regimes is highlighted by two studies. The first is by Quinlan et al. which reported that the fibrosis they observed in the hearts of aged mdx mice was patchy and occurred equally in both the left and right ventricles [51], whereas in DMD the fibrosis is predominately in the left ventricle. In contrast, the study by Nakamura et al. reported fibrotic changes in the left ventricle, and this is more comparable to that reported in DMD patients [54]. When the study designs of these two studies are analyzed it can be seen that Nakamura et al. used regular treadmill exercises for their mdx mice, which greatly increased the amount of left ventricular pathology seen, compared to the study by Quinlan et al. where the mice were not subjected to any exercise regimes.
In conclusion, the mdx mouse shares many but not all the feature of the DCM seen in DMD patients. The differences seen in the mdx mouse may be a question of scale and the size of the hearts between humans and mice and the exercise regimes used for the mdx mouse. Taking these factors into consideration the mdx mouse can provide useful information on the pathophysiology of the cardiomyopathy in DMD and with careful study design can be used to test new therapeutic targets for DMD cardiomyopathy.
As mentioned earlier both the milder BMD and DMD female carriers have also been reported to develop cardiomyopathy and to have impaired heart function. The heterozygote female mdx mice on the classic C57Bl/10 background do not develop a cardiomyopathy; even though dystrophin is expressed in only half of the cardiomyocytes, this mosaic expression of dystrophin is enough to prevent stress-induced heart damage [55]. Even by 21 months of age the hearts of the female mdx carrier mice do not display any cardiac phenotype and all physiological parameters were within the normal range whereas the hearts in the homozygote mdx by this time are displaying significant cardiomyopathic changes [56, 57]. It was also noted that utrophin was upregulated in the hearts of the female mdx carrier mice and this may compensate for the reduced dystrophin in the cardiomyocytes [58]. This study also suggests that an almost complete prevention of the DMD cardiomyopathy may only require a 30-50% mosaic dystrophin expression; this is encouraging as 100% re-expression of dystrophin is not feasible by any gene and or cell based therapy at the moment.
MDX/UTROPHIN DOUBLE DEFICIENT MICE AND CARDIOMYOPATHY
Utrophin is a related protein to dystrophin and is developmentally expressed with the same distribution as dystrophin. It is downregulated as dystrophin begins to be expressed and begins to replace it. In the adult, utrophin is only found at the neuromuscular junction where it helps to stabilize the junctional membrane. The loss of utrophin has no effect on the phenotype of mice in isolation, as dystrophin is the major structural protein of the muscle fibres. Utrophin is upregulated in the mdx mouse and its expression is located around the sarcolemma suggesting that it may partially compensate for the loss of dystrophin and perhaps to some extent explains the milder phenotype of the mdx mouse. The DKO mice that have no dystrophin or utrophin have a much more severe phenotype than the mdx mouse. They show all the clinical symptoms of DMD and the mice normally die by 20 weeks of age [59, 60]. Although the DKO mice do not die from heart failure it has been shown that there is inflammation and cardiomyocyte degeneration and fibrosis in the hearts of the double mdx/utrophin knockout mice. The severity of the heart problems is of a similar level as that seen in the aged mdx mouse; the advantage is that in these DKO hearts the cardiomyopathy develops much earlier [61]. By 2 months of age the DKO mice have abnormal ECG patterns with a drastically decreased S-R wave ratio and faster heart rates whereas the same changes are not noted until 6 months of age in the mdx mouse. These ECG abnormalities along with the polyphasic R wave have been reported in 70-80% of DMD patients. There is also a significant impairment of the contractile function in the DKO compared to the mdx mouse accompanied by impaired relaxation and a depressed β-adrenergic response [62]. Whereas it appears that dystrophin is able to fully compensate for the loss of utrophin in the single utrophin knockout mouse, the opposite is not true as there is significant dysfunction in the mdx even if it takes longer to become manifest, so utrophin is not able to fully compensate for the loss of dystrophin.
In the mdx/utrophin DKO an increased expression of the α7β1 integrin is able to ameliorate the development of the muscular dystrophy [63]. In both the mdx mouse and DMD patients there are increased levels of α7β1 and this leads to the question of whether increasing the levels of α7β1 might improve the muscle pathology by helping to stabilize the muscle membrane. Increasing the α7β1 level in the mdx/utrophin DKO does help to maintain the structural integrity of the myotendinous and neuromuscular junctions. In addition, the regenerative capacity of skeletal muscle was also increased. The upregulation of α7β1 also reduced the development of the cardiomyopathy associated with this DKO. However, the transgenic α7 transcripts were not seen in the heart and thus the cardiomyopathy is a secondary manifestation of the skeletal muscle pathology [64].
While this last study suggests that at least a part of the pathophysiology of the cardiomyopathy is not directly related to the lack of dystrophin in the cardiomyocytes but is a secondary consequence of the skeletal muscle pathology, there is still considerable evidence that the lack of dystrophin has severe consequences on the function of the cardiomyocytes. Therefore, restoring the vital link between the cytoskeleton and the DGC in only 20-30% of the cardiomyocytes and skeletal muscle may be enough to prevent the development of the cardiomyopathy and muscle damage. Certainly if dystrophin is restored in around 50% of the mdx cardiomyocytes this is able to completely prevent the stress-induced cardiomyopathy and cause the return to a normal left ventricular function [55]. Therefore, there is hope that if the various gene therapy strategies that are in clinical trials at the moment can re-establish an exon skipping, truncated dystrophin, or upregulate utrophin to target the heart as well as the skeletal muscle then there is a possibility of improving the cardiac function in DMD.
MDX/MYOD DOUBLE KNOCKOUTS AND CARDIOMYOPATHY
MyoD is basic-helix-loop-helix myogenic determination transcription factor that is not expressed in the heart but is expressed in all skeletal muscle committed cells. In the MyoD knockout skeletal muscle forms normally, this is because there is compensation from the other members of the MyoD family of transcription factors that are able to substitute for the loss of MyoD. The double mdx/MyoD knockout mice show pronounced myopathic changes caused by the reduction in the skeletal muscles ability to regenerate as the satellite cells that are the resident stem cell population require MyoD for their proper differentiation [65]. As MyoD is not expressed in the heart, any myocardial changes that are seen are the result of increased muscle damage and worse muscle regeneration. These DKOs do develop a severe cardiac myopathy. When the hearts are analyzed, there are areas of fibrosis made up of necrotic myocytes and these areas are associated with hypertrophied myocytes. The areas of fibrosis were confined to the epicardial region of the left ventricle mainly, which is similar to the fibrosis seen in DMD [66]. It has also been reported that the cardiomyocyte hypertrophy seems to precede the necrosis, suggesting that the hypertrophied cardiomyocytes are more susceptible to damage.
It has been suggested that this animal model is the best and most accurate murine model for DMD that is currently available and as such, some work has started on using this model to understand the pathophysiology of the cardiomyopathy that develops in DMD. The role of p38 and JNK-1 kinases in the development of the dystrophin-associated cardiomyopathy has been suggested as an early stage change that may lead to the later cardiomyopathy [67]. Phosphorylated p38 was dramatically reduced in the hearts from 10-month-old mice where there are significant cardiomyopathic changes in the mdx/MyoD DKOs compared to the levels observed at 7 months where the hearts are still relatively undamaged. In contrast, tyrosine phosphorylation of JNK-1 was elevated in the damaged hearts of the mdx/MyoD DKOs.
FELINE MODELS OF DMD
A colony of cats that are deficient in dystrophin have been identified [68, 69]. These cats develop a hypertrophic feline muscular dystrophy (HFMD), the muscles in these cats show multifocal areas of degeneration and regeneration and there is a wide variation in the muscle fibre diameters [69, 70]. However, muscle fibrosis has not been observed in the HFMD model, whereas in DMD there is skeletal muscle atrophy and the replacement of muscle with fibrotic tissue. In addition in the HFMD cat there is severe progressive muscle fibre hypertrophy especially of the diaphragm, the reverse of what is seen in DMD patients were the diaphragm is one of the muscles that undergoes severe muscle fibre atrophy and fibrosis which leads to the development of the respiratory problems seen in DMD patients [68, 69, 71]. The HFMD cats do though develop cardiomyopathy [70]. The changes in the hearts in the HFMD cats have been reported in affected males and females and the obligate female carriers [70]. There is early onset of concentric myocardial hypertrophy which is seen in all adult cats, these lesions were predominantly in the left ventricle, interventricular septum and the papillary muscles, they are also mainly seen in the endocardium [70]. However, because there is myocardial hypertrophy clinical signs of heart failure are very rare in the HFMD model. The HFMD model is therefore not a good model system for the study of the DMD cardiomyopathy, as it does not mirror the human condition.
CANINE MODELS OF DMD AND CARDIOMYOPATHY
There are several different canine models of DMD, including the two most studied canine models: the golden retriever muscular dystrophy (GRMD) and the beagle model of canine based x-linked muscular dystrophy (CXMD). The advantage of these canine models of DMD is that they tend to display a far more severe phenotype that mirrors the human progression far better than the mdx mouse. In both the GRMD and the CXMD there is severe muscle atrophy in the limbs that lead to joint contractures and dysphagia. The GRMD shows similar ECG findings as those reported in the human DMD patients and there is a progressive cardiomyopathy that is comparable to the cardiomyopathy in DMD [28, 72-74]. This makes the GRMD a very useful model to explore the pathophysiology and testing of potential new treatment options. However, the size of the GRMD makes it difficult and expensive to maintain any colony and this restricts its use.
The beagle is a medium sized dog making the maintenance of the CXMD colony more manageable. They also manifest abnormalities on echocardiograms and the cardiac pathology can be seen by 12 months of age. The abnormal and distinct deep Q-waves in leads II, III and aVF are seen around one year of age. The deep Q-wave has been thought to be due to the myocardial fibrosis in the posterobasal region of the left ventricle, but these changes are often seen before any changes on the echocardiograms. At 6-7 months of age there are no changes seen on the echocardiograms and no alterations in the function of the left ventricle. Therefore, the cardiac phenotype of the CXMD is milder than the GRMD but it is identical [75].
The female carriers in the canine models have also been proven to mirror the human female carriers more accurately than those of the mdx mouse. Female carrier dogs have a significantly increased Q/R ration in leads V2 and V4 [72]. There were also changes in female carrier dogs noted on the echocardiograms that represent a calcified myocardium due to fibrosis of the myocardium and replacement of the cardiomyocytes with dense connective tissue [72]. Therefore, both the canine models and their female carriers are a more accurate model for the cardiomyopathy that develops in the human DMD patients and the human female carriers.
NEW TREATMENT OPTIONS FOR MUSCULAR DYSTROPHIES AND THE HEART
Despite the tremendous progress that has been made in the past several years in developing gene therapy that targets the skeletal muscle for treating DMD, few attempts have been made to target the cardiomyopathy that develops. One study explored the potential of engraftment of fetal cardiomyocytes in the mdx and a canine model of DMD [76]. However, the results were not encouraging as the dystrophin expression was limited to the transplanted cells. An additional problem has emerged from the targeted repair of the dystrophic skeletal muscle, as once the skeletal muscle is repaired it has a tendency to make the cardiomyopathy become more apparent and severe than in the untreated mdx mice. Once again, this highlights the secondary damage on the heart from increasing the activity and mobility of the animal, which increases the stresses on the heart, composed of cardiomyocytes that are more fragile. These studies have emphasized the need to target both the skeletal and cardiac muscles to have an acceptable clinical outcome.
The present cell based therapies are based around the use of mesangioblasts rather than the satellite cells of the skeletal muscle. Mesangioblasts have several advantages over the use of satellite cells especially for the restoration of dystrophin to the heart as well as the skeletal muscle. They are multipotent progenitor cells and can thus potentially make any mesodermal tissue including skeletal and cardiac muscle. They can also be delivered via the arterial system and so can target all the skeletal muscles around the body as well as the heart [77]. These mesangioblasts have proven in the GRMD model that they are able to restore dystrophin to the skeletal muscle [78]. In a recent study a cardiac mesangioblast population has also been identified and isolated and has been shown to differentiate into cardiomyocytes when cultured in vitro and then injected into the infarcted myocardium [79]. Therefore, the principal that these mesangioblast may help to repair the cardiomyopathy in DMD has been proven. However, it has still to be shown conclusively that these cells can deliver sufficient improvement to both the skeletal and cardiac muscle functions.
The reduced number of gene therapy attempts to repair the cardiac muscle rather than the limb muscle is the result of two problems. Firstly, cardiac muscle is harder to target than the muscles of the limbs because of its anatomical location deeper in the body and because of its normal vital physiological function, as you do not want to disrupt the function of the heart during the procedure. Secondly, the size of the dystrophin cDNA exceeds the packaging capacity of the viral vectors that are currently available. This second problem has been an issue for all gene therapy treatments for DMD and has been addressed by using mini or micro-dystrophin constructs. These mini-dystrophin constructs are on average around 30% of the size of the full length dystrophin cDNA reviewed by [80]. This will produce a BMD type phenotype that is capable of providing a functional improvement in the skeletal muscles of the mdx mouse [81]. Yue et al. showed that direct cardiac injection of an adeno-associated virus (AAV) that delivered a micro-dystrophin gene to 12 hour old neonatal mdx mice was able to cause extensive expression of the micro-dystrophin in both the inner and outer layers of the myocardium even 10 months later [82]. This truncated mini-dystrophin formed a functional connection with the DGC and improved the sarcolemma integrity of the cardiomyocytes. However, the direct injection of AAV vectors carrying a mini-dystrophin is unlikely to take off and a systemic treatment option that targets the skeletal muscle and the cardiac muscle at the same time is the ideal situation.
The restoration of the open reading frame in the mutated dystrophin gene using antisense oligonucleotides, which allow exon skipping over the mutated exons and create shortened transcripts that are able to generate a truncated but functional dystrophin have shown in small scale trials to be effective for improving the dystrophin expression in skeletal muscle [83]. Systemically delivered phosphorodiamidate morpholino oligomers (PMOs) have proven to be particularly effective for skipping the mutated exon in the mdx mouse, exon 23, and producing dystrophin expression at therapeutic levels throughout the body in the skeletal muscles of the mdx mice. Unfortunately, although dystrophin could be restored in the skeletal muscles it was not detectable in the heart [84]. In a recent study the use of an arginine-rich cell-penetrating peptide, conjugated PMO has been shown to successfully restore the dystrophin expression in the heart and to significantly improve the sarcolemma integrity and prevent the development of the cardiomyopathy [85]. The treatment consisted of 2 cycles of 4 injections in each cycle (1 injection per day for 4 days) separated by a 2 week period between week 8 to 16 weeks of age in the mdx mouse. This treatment was able to prevent the development of the cardiac hypertrophy and diastolic dysfunction by 5-6 weeks after the treatment. Even with this short term treatment regime and the resultant decline in the expression of the dystrophin gene by 12 weeks after treatment had finished, to less than 1% of the original expression level, the cardiac improvement continued for up to 7 months [85, 86]. These exon-skipping trials may provide a therapeutic approach that may not only prevent but also slow down the development of the cardiomyopathy in DMD patients.
The other major treatment option is to employ molecules that allow read-through of the stop codon in the dystrophin gene that causes the loss of the dystrophin protein in DMD. Antibiotics like gentamycin have the ability to suppress stop codons and the efficiency of the read-through varies inversely to the efficiency of a stop codon. Gentamycin treatment of mdx mice was able to re-establish around 20% of the normal expression of dystrophin but this was highly variable from animal to animal and human trials using gentamycin have not proven effective. The strategy though is sound and the new drug PTC124 (Ataluren) when administered to the mdx mouse has resulted in the production of full-length functional dystrophin. It is also able to restore the dystrophin expression in the hearts of mdx mice after 4 weeks of treatment [87]. Clinical trials of PTC124 have been extended because of the positive results that have emerged so far.
New pharmaceutical treatments have also been suggested for DMD cardiomyopathy from the results obtained in the mdx mouse. One such drug that has shown beneficial effects in the mdx mouse is sildenafil. Sildenafil is a phosphodiesterase 5 (PDE5) inhibitor and prevents the breakdown of cyclic guanosine monophosphate (cGMP), sildenafil has already been shown to prevent cardiac hypertrophy and improve myocardial function in genetically normal mice that have been subjected to pressure overload [88, 89]. It has previously been shown that prior to the development of overt cardiomyopathy in the mdx mice there are metabolic and functional abnormalities, specifically there is defective nitric oxide (NO) and this has knock on consequences for its cellular effector cGMP signaling [90]. Neuronal NO synthase (nNOS) is significantly downregulated in dystrophic hearts and its normal subsarcolemma distribution is disrupted so that it is found now in the cytoplasm of the cardiomyocyte. When the expression of nNOS is restored then it has been reported that the inflammation, fibrosis and ECG abnormalities are prevented in the mdx mouse [52]. Sildenafil is able to enhance cGMP-signaling independent of NO formation; this has been reported to improve the contractile performance, metabolic status of the cardiomyocytes, and improves the sarcolemma integrity in mdx mice hearts [91, 92]. Thus, sildenafil constitutes a potential clinical pharmaceutical treatment for dystrophin related cardiomyopathies.
Another potential pharmaceutical treatment for DMD cardiomyopathy is the use of inhibitors of TGF-β. This cytokine is a likely mediator of the fibrosis that is seen in the cardiomyopathy. It is expressed at high levels in the skeletal muscles of DMD patients and its level has been shown to be related to the amount of fibrosis that develops in the skeletal muscle [93]. Pirfenidone is an orally active inhibitor of TGF-β fibroblast induced growth and collagen synthesis in a number of different tissues. It has proven useful for a number of conditions that cause fibrosis of various organs such as pulmonary fibrosis, and glomerulosclerosis in a model of renal fibrosis [94-97]. Eight-month-old mdx mice treated with pirfenidone for 7 months had lower TGF-β mRNA and displayed improved cardiac contractility that were equivalent to their wildtype littermates of the same age [98]. However, there was no change in the cardiac stiffness or the amount of fibrosis that developed in the pirfenidone treated mdx hearts [98]. The earlier administration of pirfenidone during the acute inflammatory stage might be effective at preventing the start of the fibrosis and help to maintain the function of the heart.
The loss of dystrophin from the DGC makes the cardiomyocytes more susceptible to damage and causes tears in the membrane that then allow calcium to enter the cell. The application of a membrane sealant poloxamer 188 in vitro has been shown to correct these defects [99]. In vivo administration of poloxamer 188 to mdx mice instantly improved the ventricular geometry and was able to block the development of acute cardiac failure after dobutamine mediated stress. There are still several hurdles to resolve over the optimal dosing and long-term effects of such chemical based membrane sealants in humans. However, these do represent a new and novel therapeutic approach for preventing or reversing the progression of the skeletal and cardiac muscle pathology in DMD [99-101].
CONCLUSION
The present treatment options and frequent monitoring have led to a major improvement in the quality of life of DMD patients. However, the longer life span of DMD patients has revealed that the cardiomyopathy that develops is a major health issue that needs to be addressed. The traditional view that medications for the heart should only be started when overt symptoms become evident is now being challenged, as it may already be too late to significantly improve the function of the heart by this stage. Instead, treatments are now being initiated before overt symptoms are seen to help protect the heart from damage. Because of the increasing numbers of DMD boys who are now surviving longer and developing cardiomyopathy new treatment regimes are been examined using the numerous animal models available. The best animal model to study the cardiomyopathy in DMD is still debatable; the mdx is a milder phenotype, the DKO mouse models are DKOs and may have additional deficits, and the feline model is not a good model for DMD while the canine models are not readily available and are expensive. What may help, and has helped the skeletal muscle studies, is to have a set of agreed study protocols designed to achieve the best and most reliable results in the mdx mouse and use the other models as an additional source of information. Having a set of defined study protocols means that the results from various studies can be directly compared with each other and more importantly any results can be more easily translated into modifications in the treatment regimes for the human DMD patients.
ABBREVIATIONS
ACKNOWLEDGEMENTS
LGR is supported by grants from the Muscular Dystrophy Campaign and the MRC.

