All published articles of this journal are available on ScienceDirect.

COX-2 Inhibition by Use of Rofecoxib or High Dose Aspirin Enhances ADP-Induced Platelet Aggregation in Fresh Blood
Abstract
Aim:
Increased cardiovascular risk after use of selective or nonselective cyclooxygenase-2 (COX-2)-inhibitors might partly be caused by enhanced platelet aggregability. However, an effect of COX-2 inhibition on platelets has so far not been observed in humans.
Methods:
We tested in healthy volunteers the effect of COX-2-inhibition nearly in-vivo, i.e. immediately after and even during blood sampling.
Results:
Measurement within 2 minutes after venipuncture, but not 60 minutes later, showed that 50 mg of rofecoxib (n=12) or 500 (n=8) or 1000 (n=8) mg of aspirin increased ADP-induced platelet aggregation in a whole-blood aggregometer to, respectively, 152, 176 and 204 % of basal level (p<0.01). No significant differences in aggregability were observed after ingestion of 80 mg of aspirin (n=16), or placebo (n=8). Plasma 6-keto-PGF1α was decreased to 74 % after rofecoxib and to 76 and 70 % after 500 and 1000 mg of aspirin but did not change after low dose aspirin. Continuous photometrical measurement of aggregation in blood flowing from a cannulated vein revealed that high dose aspirin did not elicit aggregation by itself, but increased ADP-induced aggregation in proportion to the decrease in prostacyclin formation (r=0.68, p = 0.004). Since in these experiments thromboxane production was virtually absent, the enhanced aggregation after partial COX-2 inhibition was not caused by unopposed thromboxane formation.
Conclusions:
We conclude that both selective and nonselective COX-2 inhibition enhances ADP-induced platelet aggregation in humans. This effect can only be detected during or immediately after venipuncture, possibly because of the short half-life of prostacyclin.
INTRODUCTION
Inhibitors of cyclooxygenase-2 (COX-2) are reported to promote cardiovascular events in patients at atherothrombotic risk. Recent studies have shown that this effect is not specific for selective COX-2 inhibitors but has been demonstrated for nonselective COX-2 inhibitors as well [1-6]. The mechanisms involved remain unclear. In rats we have found that both selective (parecoxib) and nonselective (high dose aspirin) COX-2 inhibitors enhance shear stress-induced platelet aggregation when measured nearly in-vivo, i.e. in an extracorporeal shunt between carotid and femoral artery [7]. However, the hypothesis that COX-2 inhibition increases platelet aggregability also in humans seems to be contradicted by clinical and laboratory studies [8-15].
In these studies the time between blood sampling and aggregation-testing was, as usual in the clinical setting, one hour or more. During that time the reaction of platelets to ADP and collagen increases, reaching a stabile maximum after 30 to 45 minutes [16]. This may seriously hamper and even preclude detection of an increase in platelet aggregability at one hour or more after venipuncture. Moreover, COX-2 derived endogenous platelet antagonists with a short half-life, such as prostacyclin (PGI2), which inhibits platelet aggregation with a half-time of 6 – 10 minutes in human blood and 11 to 16 minutes in human plasma [17-19], will then have lost their effect. Therefore, we compared in humans the effects of COX-2 inhibition on platelet aggregability at 2 and at 60 minutes after blood sampling, using a conventional whole-blood aggregometer. To approach the in-vivo situation even more, aggregation was in a number of experiments also measured directly in blood flowing from a cannulated vein, with aid of an established photometric device [20]. In addition, we measured circulatory levels of a stabile metabolite of PGI2 before and after COX-2 inhibition.
MATERIALS AND METHODS
Subjects and Protocol
Healthy non-smoking Caucasian volunteers (aged 20-62 years), who did not use drugs for at least two weeks before enrolment, and who did not exercise, eat fish or drink alcohol within the 12 hours before the experiment, gave informed consent. To reduce the influence of hormonal variation only male subjects were involved, and the effect of circadian variation in platelet aggregability was minimised by starting all experiments at 11.00 h am, when aggregation is maximal [21]. The effect of selective COX-2 inhibition on ADP-induced platelet aggregation was studied by testing aggregation before and 1.5 – 2 hours after intake of placebo (n=8) or 50 mg of rofecoxib (VIOXX, Merck, Sharp and Dohme, now withdrawn from the market, n=12), which is the recommended dose for relief of acute pain. It reduces COX-2 activity within 1.5 hour after ingestion for at least 10 hours [22], and has no significant effect on COX-1 activity [23]. For nonselective COX-2 inhibition high dose aspirin (Aspro, Bayer) was used in a dose of 500 mg (n=8) or 1000 mg (n=8). Aspirin was also given in low dose: 80 mg (Cardio 80 PCH, n=16). Low dose aspirin blocks COX-1 mediated platelet thromboxane-A2 (TXA2 ) formation within 20 minutes after ingestion. The blockade lasts for the life-time (approximately 10 days) of the platelets, because they lack the ability to make new protein. The inhibiting effect of low dose aspirin on COX-2 mediated prostacyclin formation in the nucleated endothelial cells is only short-lasting because of protein resynthesis, whereas high dose aspirin reduces endothelial PGI2 synthesis for more than 3 hours [24]. Platelet aggregation was elicited by ADP because ADP plays a central role in initiating and propagating aggregation induced by a variety of agonists, and because ADP-induced aggregation is known to be inhibited by PGI2 [25]. The study protocol was approved by the Medical Ethics Review Committee of our hospital.
Blood Collection
Blood was collected via an indwelling plastic cannula in an antecubital vein. Because of the possible generation of prostacyclin and other substances that affect platelet activity during cannulation, the first few milliliters were discarded. Part of the blood was added to 3.2 % sodium citrate (9:1 volume ratio) for in-vitro measurement of platelet aggregability, and another part immediately added to a solution of K3-EDTA for measurement of PGI2 levels (see Measurement of prostanoids), and for platelet and other blood cell counts (ABX Diagnostics, Netherlands).
Platelet Aggregation In-Vitro
To enable immediate measurement of aggregation in-vitro, blood was collected into a prewarmed syringe and quickly added to a whole-blood aggregometer (Chronolog Corporation, Havertown). Aggregation was elicited by 5 μM ADP within exactly 2 minutes after blood collection, and quantified as the change in impedance (ohms) at 6 minutes after addition of the agonist. Only one agonist could be tested in this way, because the aggregometer had only one channel. The unused blood was set apart at room temperature (in an air-free syringe, to prevent loss of CO2 and change of pH), and tested 60 minutes later. After the latter test the reaction to 330 μM arachidonic acid was measured as well. In a separate set of experiments (n=8) the time-dependency of the platelet reaction to 5 μM ADP was more precisely studied by varying the delay between venipuncture and in-vitro aggregation testing (between 2 and 60 minutes) in subjects who had not taken COX-inhibitors.
Platelet Aggregation in Flowing Blood
In the experiments with high dose aspirin aggregability was measured both in-vitro (see above) and nearly in-vivo by a photometric device that can detect platelet aggregates in flowing blood. For the latter measurement the venous cannula was via tubing connected to a glass capillary on the stage of a microscope. Passing platelet aggregates caused a change in light transmission, which was, after 40 x magnfication, projected on light-sensitive diodes. The rectified and integrated signal was continuously measured [20]. Blood flow through the capillary was stabilized at 2 ml/min by a small home-made roller pump. Premature platelet activation and clotting were prevented by coating the tubing with albumin and by infusing unfractionated heparin (final concentration 5 IE/ml blood) into the line before the pump.
Platelet aggregability in flowing blood was tested within 15 seconds after the blood left the body, by two-minute infusions into the tubing of incremental concentrations of ADP (10, 20, 30, 40, and 50 μM final concentration). This was followed by a two-minute infusion of 20 μM epinephrine. During platelet aggregation, induced by 30 μM ADP, blood was collected from the end of the tubing in K3-EDTA solution for assessment of plasma TXA2 (see Measurement of prostanoids).
Measurement of Prostanoids
For enzymeimmunoassays of plasma 6-keto-PGF1α and TXB2, which are the stabile metabolites of PGI2 and TXA2, respectively, EDTA-blood was centrifuged at 3500 g for 20 minutes and the platelet poor plasma stored in –80 °C until analysis by EIA Biotrak systems (Amersham Biosciences, detection limits: 3.0 and 3.6 pg/ml , respectively). Before analysis the prostaglandins were separated from fatty acids using Amprep TM minicolumns and solvent extraction, according to the manufacturer's instructions. Samples selected for measurement of 6-keto-PGF1α were divided before analysis and one part was spiked with 6-keto-PGF1α for estimation of recovery. All assays were performed in duplicate.
Statistics
Values are given as mean±standard error of the mean (SEM). The effects of selective or nonselective COX-2 inhibition on the amount of platelet aggregation at different time points (in the whole-blood aggregometer), or for several doses of ADP (in flowing blood), were analysed by two-way ANOVA for repeated measurements, followed by a Bonferroni test. Plasma levels of PGI2 and TXA2 metabolites were compared with one-way ANOVA or the paired t-test. Statistical significance was set at P<0.05.
RESULTS
Platelet Aggregation In-Vitro
Measurement of aggregation at two minutes after blood collection showed that ingestion of a single dose of 50 mg rofecoxib, in contrast to placebo, caused an increase in ADP-induced platelet aggregation (to 152 % of basal value, Fig. 1 and Table 1). This increase could not been observed at 60 minutes after venipuncture. The selectivity of rofecoxib as COX-2 inhibitor was demonstrated by an unchanged reaction to arachidonic acid (Table 1).
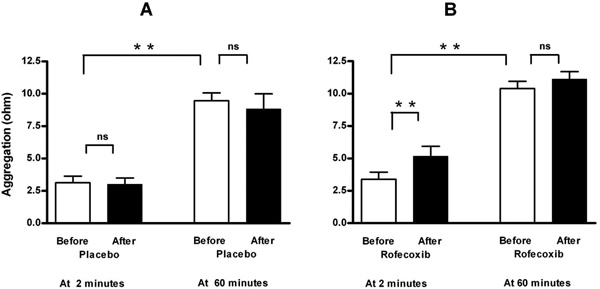
Effect of placebo (A, n=8) and selective COX-2 inhibition by a single oral dose of 50 mg rofecoxib (B, n=12) on ADP-induced platelet aggregation in a conventional whole-blood aggregometer. Enhancement by selective COX-2 inhibition is demonstrable at two minutes after blood collection, but not when the blood is set apart for 60 minutes. Means ± SEM of maximal value in ohms after application of 5 μM ADP. ** = p<0.01, ns = not significant.
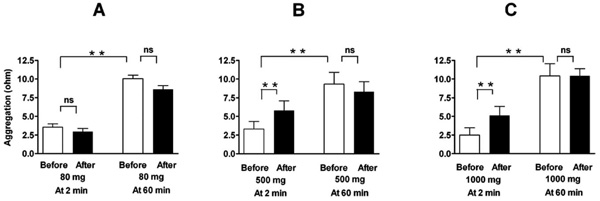
Effect of a single oral dose of 80 mg (A, n=16), 500 mg (B, n=8), and 1000 mg (C, n=8) of aspirin on ADP-induced platelet aggregation in a conventional whole-blood aggregometer. Enhancement by the higher doses of aspirin (nonselective COX-2 inhibition) is demonstrable at two minutes after blood collection, but not when the blood is set apart for 60 minutes. Means ± SEM of maximal value in ohms after application of 5 µM ADP. ** = p<0.01, ns = not significant.
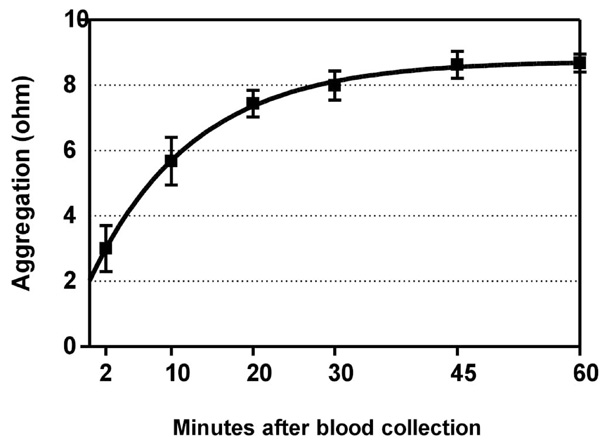
Time-dependent increase of ADP-induced platelet aggregation after venipuncture. Means ± SEM of maximal value in ohms after application of 5 µM ADP to whole blood. Half-time of the fitted curve was 8.8 minutes. Blood was collected from subjects (n=8) who had not ingested a COX-2 inhibitor, and was kept in closed tubes at room temperature.
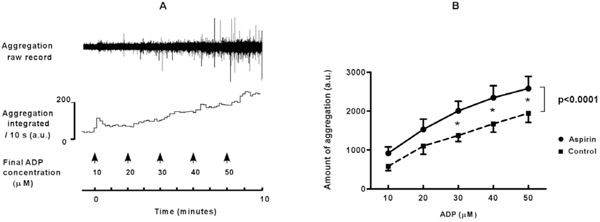
A. Platelet aggregation in flowing blood is induced by infusion of increasing concentrations of ADP into tubing connected to a cannulated vein. The aggregates were detected photometrically when they passed a glass capillary connected to the tubing. Raw signals are rectified and integrated over periods of ten seconds. A.u.: arbitrary units. B. Platelet aggregation measured directly in flowing blood is enhanced by ingestion of a high dose of aspirin (pooled data for 500 and 1000 mg). Means ± SEM represent the total amount of aggregation during two minutes of ADP-infusion, before and after high dose aspirin. * = p<0.05. Overall difference between aspirin and control: p<0.0001.
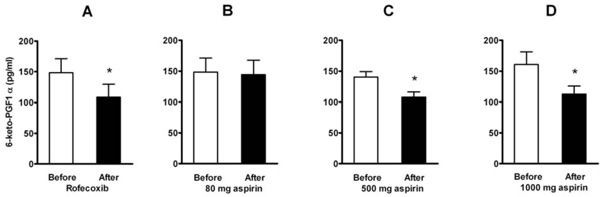
Effect of a single dose of 50 mg of rofecoxib (A, n=12) and of aspirin in doses of 80 mg (B, n=16), 500 mg (C, n=8), and 1000 mg (D, n=8) on plasma levels of 6-keto-PGF1α . Means ± SEM. * = p<0.05.
Effect of a Single Dose of Rofecoxib (50 mg) or Aspirin on In-Vitro Platelet Aggregation in a whole Blood Aggregometer at 2 and 60 Minutes after Blood Sampling
| at | Placebo | Rofecoxib | Aspirin | |||
|---|---|---|---|---|---|---|
| 80 mg | 500 mg | 1000 mg | ||||
| ADP | 2 min | c 3.1 ± 0.4 | c 3.4 ± 0.5 | c 3.5 ± 0.4 | c 3.3 ± 1.0 | c 2.5 ± 1.0 |
| 3.0 ± 0.4 | 5.2 ± 0.8 ** | 3.0 ± 0.4 | 5.8 ± 1.3 * | 5.1 ± 1.2 * | ||
| ADP | 60 min | c 9.5 ± 0.5 | c 10.4 ± 0.5 | c 10.1 ± 0.5 | c 9.3 ± 1.6 | c 10.4 ± 1.6 |
| 8.8 ± 0.9 | 11.1 ± 0.6 | 8.6 ± 0.5 | 8.3 ± 1.4 | 10.4 ± 1.0 | ||
| Arach. Acid | 60 min | c 10.4 ± 0.5 | c 10.8 ± 0.6 | c 10.9 ± 0.7 | c 11.5 ± 0.9 | c 9.9 ± 1.2 |
| 9.4 ± 0.6 | 11.3 ± 0.7 | 1.1 ± 0.3 ** | 1.1 ± 0.7 ** | 0.2 ± 0.2 ** | ||
| n | 8 | 12 | 16 | 8 | 8 | |
Maximal reactions to ADP (5µM) and arachidonic acid (333μM) in ohms (means ± SEM). c = control condition before ingestion of the study drug.
* = p<0.05 and
** = p<0.01, compared to control.
Literature Data on Mean Basal Levels of Plasma 6-keto-PGF1α
| Method | Subjects | Mean Plasma 6-keto-PGF1α (pg/ml) | Ref. |
|---|---|---|---|
| RIA | healthy adults | 291, 245, 182, 161, 126, 115, 110, 88, 85, 26 | [i-x] |
| patients before surgery | 225, 154, 96, 53, 97, 46, 72, 109, 120 | [xi-xix] | |
| RIA after separation on charcoal or HPLC | healthy adults | <7.5, 4.7, 1.7, 1 | [xx-xxiii] |
| ELISA | healthy adults | 165, 297, 149, 145, 39, 38 | [xxiv-xxix] |
| patients before surgery | 200, 57 | [xxx, xxxi] |
RIA: radioimmunoassay. HPLC: High Performance Liquid Chromatography. ELISA: Enzyme-Linked Immuno Sorbent Assay. For references see APPENDIX
Comparable results were found with nonselective COX-inhibition by high dose aspirin. Whereas low dose aspirin (80 mg) had no effect, higher doses of aspirin (500 and 1000 mg) elicited a similar increase in aggregation as rofecoxib did (Fig. 2 and Table 1). This was, again, demonstrable only when aggregation was measured immediately after blood sampling, but not when the blood was set apart and aggregation elicited 60 minutes later. Reactions to arachidonic acid (measured only after more than 60 minutes) were suppressed by all doses of aspirin (Table 1).
Time-dependency of the in-vitro platelet reaction to ADP in subjects who had not taken COX-inhibitors is depicted in Fig. (3). Platelet aggregability (y) increased with the delay between venipuncture and aggregation testing (x) according to y= 8.75 - 6.73·e -0.079·x , and reached a plateau at about 45 minutes. The time to reach half the maximal value was 8.8 minutes. The level at 60 minutes was similar for arachidonic- and for ADP-induced aggregation (Table 1), suggesting a maximal response, and illustrating the necessity to measure a possible increase in aggregability not at 60 minutes but quickly after venipuncture.
Platelet Aggregation in Flowing Blood
The rather unexpected findings with high dose aspirin were corroborated by testing platelet aggregability directly in blood flowing from a cannulated vein. In unstimulated blood no spontaneous platelet aggregation was detected, neither in control nor after aspirin ingestion. This indicates that high dose aspirin by itself did not elicit aggregation. In accordance, platelet counts were not significantly diminished (261 ± 14 x 109/L versus 275 ± 12 x 109/L in control), and other cell counts remained within normal ranges (not shown). Addition of ADP elicited a dose-dependent increase in aggregation (for an example see Fig. 4A). These responses were enhanced to 160 ± 20 % of control ((p<0.0001, Fig. 4B) after ingestion of high dose aspirin, without a significant difference between experiments with 500 or with 1000 mg of aspirin. This enhancement was unlikely caused by unopposed TXA2 activity, because TXA2-generation during ADP-induced platelet aggregation in flowing blood was markedly reduced after aspirin (from 552 ± 96 to 30 ± 7 pg/ml, p < 0.0001).
Also reactions of the platelets to 20 μM of epinephrine, injected after the series of ADP additions, tended to be stronger after high dose aspirin (146 ± 22 % of control, p=0.09).
Prostacyclin
Basal levels of plasma 6-keto-PGF1α , the stabile metabolite of prostacyclin, were reduced after a single dose of rofecoxib (to 74 %) and after the higher doses of aspirin (to 76 % after 500 mg and to 70 % after 1000 mg), but not after low dose aspirin (to 98 %) (Fig. 5). When aggregation was tested in flowing blood, a positive correlation (Pearson r=0.68, p = 0.004) was found between the individual amount of 6-keto-PGF1α reduction and the aspirin-induced increase of platelet aggregation (averaged for the five doses of ADP).
DISCUSSION
This study shows that both selective and nonselective COX-2 inhibition by, respectively, rofecoxib or high dose aspirin enhances ADP-induced platelet aggregation in healthy humans. This enhancement was demonstrable only with near-in-vivo measurement of platelet aggregation in blood flowing from a cannulated vein, or in-vitro with a whole blood aggregometer, if aggregation was tested within the first minutes after blood sampling, but not if the blood was set apart and the measurements postponed for one hour. The present study extends previous findings of increased platelet aggregability after COX-2 inhibition in rats [7], and is in line with studies showing increased platelet adhesion in hamster arterioles [26] and rabbit injured carotid arteries [27], after COX-2 inhibition.
COX-2 Inhibition and Increased Thrombotic Risk
During measurement of platelet aggregation in blood flowing from a cannulated vein no aggregates were detected as long as platelets were not stimulated by an exogenous agonist, neither in control nor after partial COX-2 inhibition by aspirin. Therefore, COX-2 inhibition did not by itself elicit platelet aggregation. This suggests that inhibition of COX-2 in healthy man is of limited risk. In cardiovascular patients, however, COX-2 inhibition might amplify aggregation elicited by e.g. rupture of an atherosclerotic plaque or further narrowing of a vascular stenosis.
The results with an adult dose of aspirin (500-1000 mg) contrasts to the well known beneficial effect of low dose aspirin in preventing restenosis. It might, however, explain why in the Cottbus study significantly more reinfarctions were seen with 1000 mg of aspirin per day than with 30 or 60 mg/day [28]. Also more recent studies showed that the risk of stroke, myocardial infarction, and death was significantly greater for patients taking higher doses of aspirin (650-1300 mg) compared to those taking lower doses (81-325 mg) [29-32]. The results with high dose aspirin show at the same time that it is not the selectivity of COX-2 inhibitors that makes them prothrombotic. This could explain why cardiovascular risk is also increased by use of nonselective COX inhibitors like ibuprofen and diclofenac (hazard ratios of, respectively, 1.51 and 1.63 [2-6]).
Time-Dependency of Platelet Aggregation
Our near in-vivo measurements explain why the stimulating effect of COX-2 inhibition on platelet aggregation has not been detected in clinical settings where measurements are usually delayed by transport of the blood samples to a local laboratory [8-15]. Hence, our findings show that important information about the thrombotic risk of a patient may be missed when aggregation is not measured immediately after venipuncture.
A time-dependent increase of platelet aggregability after venipuncture (see our Fig. 3) has been found earlier by other investigators using whole blood analysers [16] or flow cytometry [33], indicating that the method used is irrelevant. Inoue et al. [34], using the screen filtration method, found that an enhancing effect of cigarette smoking on platelet aggregation was demonstrable only at 5 minutes, but not at 60 minutes. These findings are compatible with our results, particularly because smoking is, just as COX-2 inhibition, known to inhibit PGI2 formation [35].
The mechanism for the time-dependent increase in platelet aggregability after venipuncture awaits to be clarified. It has been postulated that the increase is caused by a temperature-dependent change in the intra-platelet calcium level [16], or by degradation of labile platelet inhibitors like PGI2, EDRF/NO and ecto-ADPases.
Thromboxane A2 is not Essential for the Enhancement of ADP-Induced Platelet Aggregation by COX-2 Inhibition
A common hypothesis states that selective COX-2 inhibitors might be prothrombotic by inhibiting prostacyclin formation while leaving COX-1 induced thromboxane formation unopposed, in this way disturbing the balance between suppression and promotion of aggregation [26]. In our experiments with high dose aspirin, however, thromboxane formation was nearly abolished. Hence, the increase of ADP-induced aggregation in these experiments was not an effect of unopposed TXA2 formation. This might be explained by the fact that thromboxane plays a minor role only in ADP- induced aggregation [36, 37]. Although in the clinical situation also TXA2 –dependent agonists will be involved, various studies showed that the risk of myocardial infarction was not reduced when the use of a selective COX-2 inhibitor was combined with low dose aspirin [5, 38].
Possible Role of Prostacyclin
Selective or nonselective COX-2 inhibition by a single dose of respectively rofecoxib or high dose aspirin reduced the plasma PGI2 levels by 24 to 30 %. Although this reduction may seem moderate, ADP-induced platelet aggregation was enhanced by 52 to 60 %. It might be speculated that this enhancement will be even stronger with daily use of COX-2 inhibitors. Measurement of aggregation in blood flowing from a cannulated vein furthermore revealed a significant correlation between the increase in aggregation and the degree of PGI2 reduction. These results give for the first time direct evidence for the hypothesis of Moncada and Vane [39], i.e. that the basal level of circulating endogenous PGI2 is sufficiently high to partially depress platelet aggregation in human blood. This hypothesis was rejected in earlier studies, with the argument that the PGI2-levels in circulating blood are too low to influence platelet aggregability [40]. Table 2 shows, however, a number of studies in which levels of PGI 2 were found comparable to those found in the present study.
The inhibiting influence of PGI2 on platelets has been thought to occur via stimulation of platelet adenylate cyclase and elevation of platelet cAMP, which reduces intracellular levels of free calcium [41-43].
Our finding that COX-2 inhibition did not by itself elicit platelet aggregation complies with the absence of spontaneous thrombosis in mice lacking receptors for PGI2 [44], and the observation of Arehart et al. [45] in patients with a naturally occurring mutation in the prostacyclin receptor. These authors conclude that a deficiency in prostacyclin signaling is not likely an initiating factor in cardiovascular disease but accelerates the course of disease in those patients with the greatest risk factors.
Reduction of PGI2 synthesis is not exclusively caused by use of selective or nonselective COX-2 inhibitors, or by smoking, but may also occur in several diseases, including diabetes [46], or might result from use of medicines, e.g. inhibitors of vascular endothelial growth factor (VEGF) in cancer patients [47]. In addition, plasmatic half-life of PGI2 is significantly shortened during severe infections, and in acute myocardial infarction [17, 48], and in general, by lowered levels of HDL-c and apoA1, the latter being described as a potential PGI2-stabilising factor [17]. Our findings suggest that in all such conditions important information about the thrombophilic status of a patient will be overlooked if platelet aggregability is not tested immediately after blood collection ("near patient measurement").
STUDY LIMITATIONS
The results of this study are based on experiments with a limited number of healthy non-smoking male subjects under controlled conditions. They did not use drugs for at least two weeks before enrolment, and did not exercise, eat fish or drink alcohol within the 12 hours before the experiment, and the effect of circadian variation in platelet aggregability was minimised by starting all experiments at 11.00 h am. It remains to be demonstrated that the enhancing effect of COX-2 inhibition on platelet aggregability is also present in women, and in less controlled conditions.
To demonstrate that the effect of COX-2 inhibition on platelet aggregability in-vitro can only be observed when platelet aggregability is tested immediately after blood collection, only one agonist (ADP) was used, because our aggregometer had only one channel. The experiments might be repeated with other platelet agonists.
CONCLUSION
Reduction of endogenous PGI2 by intake of selective or nonselective COX-2 inhibitors enhances ADP-induced platelet aggregation in humans. This can be demonstrated when aggregation is measured during the first minutes after blood withdrawal, but not one hour later. The results imply, first, that the level of endogenous PGI2 is sufficiently high to continuously reduce platelet aggregation under basal conditions; second, that the enhancement of aggregability by COX-2 inhibition does not necessarily depend on increased TXA2 activity; and, third, that important information about the thrombotic risk of a patient may be missed when aggregation is not measured immediately after venipuncture. Our results underline the concept that use of COX-2 inhibitors increases the risk for cardiovascular events by creating a prothrombotic environment.
CONFLICT OF INTEREST STATEMENT
We declare that we have no conflict of interest.
ACKNOWLEDGEMENTS
We thank M.H. van Wijhe for the measurement of 6-keto-Prostaglandin F1α and Thromboxane B2, and Prof. Dr. V.W. M. van Hinsbergh for critical reading of the manuscript.

