All published articles of this journal are available on ScienceDirect.

Involvement of Signaling Molecules on Na+/H+ Exchanger-1 Activity in Human Monocytes
Authors Info & Affiliations
Abstract
Background:
Sodium/hydrogen exchanger-1 (NHE-1) contributes to maintaining intracellular pH (pHi). We assessed the effect of glucose, insulin, leptin and adrenaline on NHE-1 activity in human monocytes in vitro. These cells play a role in atherogenesis and disturbances in the hormones evaluated are associated with obesity and diabetes.
Methods and Results:
Monocytes were isolated from 16 healthy obese and 10 lean healthy subjects. NHE-1 activity was estimated by measuring pHi with a fluorescent dye. pHi was assessed pre- and post-incubation with glucose, insulin, leptin and adrenaline. Experiments were repeated after adding a NHE-1 inhibitor (cariporide) or an inhibitor of protein kinase C (PKC), nitric oxide synthase (NOS), nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, phosphoinositide 3-kinases (PI3K) or actin polymerization. Within the whole study population, glucose enhanced NHE-1 activity by a processes involving PKC, NOS, PI3K and actin polymerization (p = 0.0006 to 0.01). Insulin-mediated activation of NHE-1 (p = <0.0001 to 0.02) required the classical isoforms of PKC, NOS, NADPH oxidase and PI3K. Leptin increased NHE-1 activity (p = 0.0004 to 0.04) through the involvement of PKC and actin polymerization. Adrenaline activated NHE-1 (p = <0.0001 to 0.01) by a process involving the classical isoforms of PKC, NOS and actin polymerization. There were also some differences in responses when lean and obese subjects were compared. Incubation with cariporide attenuated the observed increase in NHE-1 activity.
Conclusions:
Selective inhibition of NHE-1 in monocytes could become a target for drug action in atherosclerotic vascular disease.
INTRODUCTION
The sodium/hydrogen exchanger-1 (NHE-1) is a ubiquitous integral membrane protein expressed in mammalian cells [1]. Its main role is intracellular pH (pHi) maintenance, achieved by exchanging 1 intracellular H+ for 1 extracellular Na+ [1]. It is also important in cell volume maintenance and cytoskeletal reorganization. Furthermore, it takes part in cell proliferation, apoptosis and migration [1]. NHE-1 is stimulated by intracellular acidosis, hormones and growth factors and inhibited by amiloride derivatives [2].
The role of NHE-1 inhibitors in cardiovascular disease has been investigated during the past decade. In animal models, cariporide in combination with metoprolol decreased myocardial infarction size [3]. However, human clinical trials did not show clinical benefit of NHE-1 inhibition [4].
NHE-1 overactivity is documented in overweight and obese subjects and correlated well with body mass index (BMI) but not with plasma insulin levels [5]. In obese animal models inhibition of NHE-1 improved insulin sensitivity and endothelial function [6]. Furthermore, human erythrocytes obtained from obese subjects NHE-1 were activated by insulin [7], leptin [8] and adrenaline [9] and by glucose [10] in healthy individuals. In monocytes obtained from healthy subjects, NHE-1 was activated by glucose [11] and leptin [12]. However, the influence of obesity on NHE-1 activity and its signaling pathway in human monocytes remains poorly documented. NHE-1 is involved in monocyte adhesion, migration and oxidized low density lipoprotein (oxLDL) phagocytosis under the influence of mediators such as glucose, insulin, leptin and adrenaline [13-17]. In addition to obesity, NHE-1 activation could contribute to the increased cardiovascular risk associated with insulin resistance and type 2 diabetes mellitus [18, 19].
We investigated the effect of high concentrations of glucose, insulin, leptin and adrenaline on the activity of NHE-1 by measuring the pHi in monocytes obtained from lean and obese healthy subjects. Furthermore, we investigated the role of protein kinase C (PKC), nitric oxide (NO) synthase (NOS), nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, phosphoinositide 3-kinases (PI3K) and actin polymerization on NHE-1 activity.
MATERIALS AND METHODS
Materials
Source of reagents were described elsewhere [13, 14] apart from the following: 2',7'-bis-(carboxyethyl)-5(6)-carboxyfluoresceinacetoxymethyl ester (BCECF/AM) was purchased from AppliChem (Darmstadt, Hesse, Germany). 4,4΄-di-isothiocyanato stilbene-2,2΄-disulfonic acid (DIDS), nigericin, methazolamide, iodoacetic acid, DPI (diphenyleneiodonium chloride), L-NAME (Nω-Nitro-L_arginine methyl ester hydrochloride) were obtained from Sigma (St. Louis, MO, USA). GF109203X and Gö6976 were purchased by Alexis (Lausen, Switzerland). Cytochalasin-D was obtained by Fluka (Seelze, Germany). All other reagents were of analytical grade and were obtained from commercial sources.
Subjects
Healthy obese [n = 16; BMI ≥30 Kg/m2] subjects aged between 18-35 years (13 female) attending the obesity outpatient clinic and 10 healthy lean (BMI <25 Kg/m2) age-matched subjects from the hospital staff (8 female), were enrolled in the study. None of the participants were taking any medication. All participants gave their written informed consent in accordance with the Declaration of Helsinki. This patient population was used in a previous study to assess the effect of leptin, adrenaline, insulin and glucose on monocyte function (adhesion, migration, CD36 expression, oxidized low density lipoproteins phagocytosis) [13, 14, 17].
Anthropometric measurements (body weight, height and waist circumference) and blood pressure were recorded by the same examiner and were reported elsewhere [13, 14, 17].
Study Protocol
Blood was collected after an overnight fast and distributed as previously described [13, 14, 17]. Measurement of pHi (as an estimate of NHE-1 activity) was assayed in isolated monocytes pre- and post- incubation with glucose, insulin, leptin or adrenaline. The glucose (20 mmol/l), insulin (50 μU/ml), leptin (160 ng/ml) and adrenaline (520 pmol/l) concentrations used were similar to those in previous studies [11, 13, 14, 16, 17]. All experiments were repeated after adding cariporide, a NHE-1 inhibitor. In order to investigate the signaling molecules involved the experiments were repeated after adding inhibitors of classical isoforms of PKC (Gö6976), all isoforms of PKC (GF10923X), NOS (L-NAME), NADPH oxidase (DPI), PI3K (wortmannin) and actin polymerization (cytochalasin-D).
Each experiment included 10 lean subjects and 12 obese subjects. The 12 obese subjects were randomly selected from the 16 available for each experiment. In the obese group, 8 subjects were insulin sensitive and 8 were insulin resistant as defined by euglycemic hyperinsulinemic clamp [14]. In order to investigate the influence of obesity, we performed a subgroup analysis of the lean (n = 10) and obese (n = 12) subjects.
Monocyte Isolation
Monocytes were isolated as previously described [13, 14] (Ficoll-Paque Plus followed by 46% iso-osmotic Percoll in CM). Monocyte purity measured using a Beckman Coulter (Brea, California, USA) EPICS XL-MCL flow cytometer and CD14 antibody was > 85%.
Measurement of pHi
pHi was measured using BCECF-AM [10, 11, 20]. Monocytes were washed 3 times with PBS (phosphate buffer solution) and then suspended in HCO3-free buffer NaCl (135 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 5 mM glucose, 20 mM HEPES; pH 7.3) to deplete them of HCO3-. This buffer was used for all determinations of pH unless otherwise stated. The monocytes were then loaded with BCECF-AM (1 mg/ml per 106 cells) and incubated for 30 min at 37oC in the dark. After incubation with the fluorescent dye monocytes were washed 5 times at room temperature at 1500 rpm with NaCl medium in order to remove the unbound fluorescent dye and suspended to the desired concentration (106 cells/ml). DIDS (0.125 mM) and methazolamide (0.4 mM) were added in order to avoid HCO3-/Cl- anion exchanger interference. Iodoxic Na (1 mM) was added to suppress glycolysis. The 10 μl of the inhibitor [Gö6976 (500 nM) inhibits α, β and γ isoforms of PKC, GF109203X (10 μM) inhibits all isoforms of the PKC, L-NAME (100 μM) inhibits NOS, DPI (10 μM) inhibits NADPH oxidase, wortmannin (50 nM) inhibits PI3K, cytochalasin D (2 μM) inhibits actin polymerization, cariporide (20 nM) inhibits the action of NHE-1] were added, when appropriate. An incubation for 30 min at 37oC in the dark followed. Then 10 μl of the mediactors [glucose (20 mmol/l), insulin (50 μU/ml), leptin (160 ng/ml) and adrenaline (520 pmol/l)] were added and incubated for 15 min at 37oC. Fluorescence was measured in a FL WINLab luminescence spectrometer (PerkinElmer, Waltham, MA, USA) in a 96-well black polystyrene plate with excitation and emission wavelengths set at 495 and 530 nm respectively, using 2.5 nm slit. Routinely, fluorescence was also measured with excitation wavelength set at 440 nm. At this wavelength fluorescence is proportional to intracellular dye concentration and is relatively pH-insensitive. Data were obtained as the ratio of the fluorescence at pH-sensitive excitation wavelength 495 nm and the fluorescence at the pH-insensitive excitation wavelength 440 nm.
Routinely calibration of fluorescence to pH was carried out by suspending the cells in K+ solutions (KCl 130 nM, MgCl2 1 mM, HEPES 30 mM) at 3 different pH values (6.7, 7 and 7.3) [21].The concentration of cells was 106 cells/ml. The cells were then washed with the same buffer and nigericin (13 μM) was added to each well. Nigericin promotes ion-channel opening equalizing pHi to the extracellular pH. Fluorescence was measured 5 min after nigericin addition. The curve obtained between the ratio of fluorescence and pH was linear. The pH of each solution was measured by a pH meter (Accumet, Fischer Scientific, Hampton, New Hampshire, USA).
Statistical Analysis
Statistical analysis was performed using SPSS v 15.0 for Windows (SPSS Inc., Chicago, Illinois). All values are expressed as mean ± standard deviation (SD), unless otherwise noted. Normality was tested with the Shapiro-Wilk test. Differences in continuous variables were assessed by means of 1-way analysis of variance, Student’s t-test or paired t-test where appropriate. When necessary, non-parametric tests were used. A 2-tailed p < 0.05 was considered significant.
RESULTS
Anthropometric Characteristics
As previously described [13, 14], the mean age of the subjects was 29 ± 3 years; there was no difference between the lean (n = 10) and the obese group (n = 16) (p > 0.1). The BMI was 23.8 ± 0.9 kg/m2 for the lean and 37.9 ± 7.1 kg/m2 for the obese subjects. The mean waist circumference was 86 ± 2 cm for the lean and 112 ± 13 cm for the obese subjects.
Measurement of pHi
Incubation with Glucose in the Overall Population
Glucose significantly increased pHi (p <0.0001). Cariporide inhibited glucose-induced increase in pHi (p = 0.003). Monocyte incubation with Gö6976, GF109203X, L-NAME, wortmannin or cytochalasin-D inhibited the glucose-induced increase in pHi (p = 0.008, p = 0.002, p = 0.0006, p= 0.01 and p = 0.001, respectively). The addition of DPI inhibited the glucose-induced increase in pHi but this was not significant (p = 0.056).
Incubation with Glucose: Subgroup Analysis, Lean and Obese
Glucose significantly increased pHi in both groups (p = 0.003 in lean and p = 0.006 in obese subjects). Cariporide inhibited glucose-induced increase in pHi in both groups (p = 0.005 in lean and p = 0.03 in obese subjects). Monocyte incubation with GF109203X, L-NAME or cytochalasin-D inhibited the glucose-induced increase in pHi in both groups (p <0.04). However, after adding Gö6976 or wortmannin the glucose-induced increase in pHi was significantly attenuated only in lean subjects (p = 0.008 and p = 0.034, respectively). The addition of DPI inhibited the glucose-induced increase in pHi only in obese subjects but this was not significant (p = 0.061).
Incubation with Insulin in the Overall Population
Insulin significantly increased pHi (p <0.0001). Cariporide inhibited insulin-induced increase in pHi (p = 0.002). Monocyte incubation with Gö6976, L-NAME, DPI or wortmannin inhibited insulin-induced increase in pHi (p = 0.002, p = 0.008, p = 0.02 and p = 0.005, respectively). Cytochalasin-D inhibited insulin-induced increase in pHi but this was not significant (p = 0.07). The addition of GF109203X had no effect on pHi.
Incubation with Insulin: Subgroup Analysis, Lean and Obese
Insulin significantly increased pHi in both groups (p = 0.02 in lean and p = 0.003 in obese subjects). Cariporide inhibited insulin-induced increase in pHi in both groups (p = 0.03 in lean and p = 0.039 in obese subjects). Monocyte incubation with Gö6976 or L-NAME inhibited the insulin-induced increase in pHi in both groups (p <0.05). After adding wortmannin insulin-induced increase in pHi was significantly attenuated only in lean subjects (p = 0.04). Incubation with DPI inhibited insulin-induced increase in pHi only in obese subjects (p = 0.037). The addition of GF109203X or cytochalasin-D had no effect on pHi.
Incubation with Leptin in the Overall Population
Leptin significantly increased pHi (p = 0.0004). Cariporide inhibited leptin-induced increase in pHi (p = 0.001). Monocyte incubation with Gö6976 or cytochalasin-D inhibited leptin-induced increase in pHi (p = 0.003 and p = 0.04, respectively). The addition of GF109203X, L-NAME, DPI or wortmannin had no effect on pHi.
Incubation with Leptin: Subgroup Analysis, Lean and Obese
Leptin significantly increased pHi in both groups (p = 0.016 in lean and p = 0.04 in obese subjects). Cariporide inhibited leptin-induced increase in pHi in both groups (p = 0.03 in lean and p = 0.02 in obese subjects). Monocyte incubation with Gö6976 inhibited leptin-induced increase in pHi in both groups (p = 0.001 in lean and p = 0.033 in obese subjects). However, after adding cytochalasin-D leptin-induced increase in pHi was attenuated only in obese subjects (p = 0.032). The addition of GF109203X or L-NAME did not significantly inhibit leptin-induced increase in pHi only in obese subjects (p = 0.052 and p = 0.058, respectively). The addition of DPI or wortmannin had no effect on pHi.
Incubation with Adrenaline in the Overall Population
Adrenaline significantly increased pHi (p <0.0001). Cariporide inhibited adrenaline-induced increase in pHi (p = 0.0004). Monocyte incubation with Gö6976, L-NAME or cytochalasin-D inhibited adrenaline-induced increase in (p = 0.001, p = 0.01 and p = 0.01, respectively). The addition of GF109203X, DPI or wortmannin had no effect on pHi.
Incubation with Adrenaline: Subgroup Analysis, Lean and Obese
Adrenaline significantly increased pHi in both groups (p = 0.006). Cariporide inhibited adrenaline-induced increase in pHi in both groups (p = 0.01 in lean and p = 0.047 in obese subjects). Monocyte incubation with Gö6976 inhibited adrenaline-induced increase in pHi in both groups (p = 0.024 in lean and p = 0.017 in obese subjects). However, adding L-NAME, wortmannin or cytochalasin-D significantly attenuated adrenaline-induced increase in pHi only in lean subjects (p = 0.004, p = 0.003 and p = 0.031, respectively). The addition of GF109203X or DPI had no effect on pHi.
DISCUSSION
Similar to our findings, high concentrations of glucose increased NHE-1 activity in human umbilical vein endothelial cells [22] and in human monocytes [11]. There is evidence that a higher level of glucose (5 vs 20 mM) results in significantly (p < 0.001) greater increase in pHi in monocytes obtained from healthy subjects [11]. This effect was assessed by inhibition of ethylisopropyl amiloride (EIPA) [11]. Furthermore, glucose induced hypertrophy of cardiomyocytes through the involvement of NHE-1 [23].
Inhibition of NHE-1 prevented insulin-induced glucose uptake by rat ventricular cardiomyocytes [24]. In contrast, in insulin resistant whole animal models cariporide improved insulin sensitivity [6]. Furthermore, insulin phosphorylated NHE-1 in 3T3-L1 adipocytes [25] and increased its activity in cardiomyocytes [24] and in human erythrocytes [7]. A controversial finding was reported in vascular endothelial and smooth muscle cells, where insulin inhibited NHE (mainly NHE-1) activity [26]. In our study, insulin activated NHE-1 and was inhibited by cariporide.
Leptin increased NHE-1 activity, a finding supported by previous studies in human erythrocytes [8]. In human monocytes obtained from healthy donors, leptin-induced increase in adhesion, migration, CD36 expression and oxLDL phagocytosis was mediated by NHE-1 (i.e. inhibited by cariporide) [16]. In the latter study, the role of several signaling molecules was also assessed. However, pHi was not measured. In another study, leptin-induced increase in pHi showed a dose-response pattern [12]. However, the role of signaling molecules was not assessed [12].
Another hormone that activated NHE-1 is adrenaline, which was also reported to increase NHE-1 activity in erythrocytes [9] and in cardiomyocytes [27].
We have previously shown that cariporide (a NHE-1 inhibitor) eliminated leptin-, adrenaline- glucose- and insulin-induced increase in adhesion, migration of monocytes, CD36 expression and monocyte phagocytosis of oxidized-LDL in some patient groups [11, 13-17]. Furthermore, cariporide may exert anti-atherogenic effects [15, 28] by inhibiting monocyte adhesion and expression of intercellular adhesion molecule (ICAM) [29]. The potential effect of cariporide on monocytes could be mediated through NHE-1 inhibition [13, 14, 17] or another action. However, these promising experimental results were not supported by the findings of a clinical trial. In the EXPEDITION (for Na+/H+ Exchange inhibition to Prevent coronary Events in acute cardiac condition) trial cariporide administration to coronary artery bypass graft patients (n = 5761) resulted in significantly less death + myocardial infarction compared with the placebo group (20.3% vs 16.6%; p = 0.0002) [4]. However, there was a significant increase in mortality mainly due to cerebrovascular events [1.5% in the placebo group vs 2.2% with cariporide (p = 0.02)] [4]. Therefore, it is unlikely that cariporide will be further investigated it is unlikely that cariporide will be further investigated [4].
We found that PKC and NOS were involved in the NHE-1 signaling pathway in all subjects. Similarly, PKC inhibition decreased NHE-1activity in bovine neutrophils [30]. Furthermore, in animal cardiomyocytes, PKC was involved in NHE-1 inhibition [31]. Ex vivo studies in human monocytes and erythrocytes indicate an involvement of PKC in NHE-1 activation by glucose [10] and insulin [7]. An interaction between NOS and NHE-1 is supported by a previous animal study where inhibition of NHE-1 resulted in decreased activity of neuronal NOS [32]. In contrast, inhibition of NOS was associated with increased NHE-3 mRNA and protein in other cells [33]. However, monocytes do not express NHE-3 [34]. Therefore, different isoforms of NHE may be inhibited/stimulated by the same mediator. The response may also depend on the location of the NHE.
PI3K was involved in NHE-1 activation by glucose, insulin and adrenaline only in lean subjects which could reflect a dysfunction of this kinase in the obese subjects. PI3K was involved in NHE-1 activation pathway in monocytes [16] and its activity was decreased in human skeletal muscle cells from insulin resistant obese subjects [35]. Furthermore, there is a regulatory disruption between the subunits of PI3K in obesity resulting in alteration of PI3K function [36]. The disrupted function of PI3K was also found in animal models of diet-induced obesity [37]. Alternatively, our findings may reflect the low numbers of subjects in the subgroup analysis.
Actin polymerization is involved in NHE-1 activation by leptin. The interaction of NHE-1 with cytoskeleton is well established [38] and seems to be bidirectional. NHE-1 is important for de novo assembly of actin filaments [39, 40] and its activation may contribute to actin-filaments organization [41, 42]. On the other hand, NHE-1 localization in the plasma membrane requires an intact actin cytoskeleton [43]. The cytoskeleton may differ in obese subjects since in animal models a high fat meal decreased the mRNA of cytoskeleton [44]. Furthermore, obesity commonly coexists with insulin resistance and hyperinsulinemia. Altered structure of F-actin was found in insulin resistance [45] and exposure to insulin led to actin filament rearrangement in skeletal muscle cells [46] and 3T3-L1 adipocytes [47].
Leptin is another hormone that is increased in obesity and it influences actin cytoskeletal dynamics in hippocampal neurons [48] and in vascular tissue [49]. It is therefore of interest that in our subgroup analysis, the leptin-induced increase in NHE-1 activity was reduced by cytochalasin-D only in the obese patients.
NADPH oxidase is involved in NHE-1 activation by insulin. It is known that NHE-1 and NADPH oxidase interact in a reciprocal way. NADPH oxidase stimulation produces acid thus activating NHE-1 which alkalinizes the pH [50]. Furthermore, in animal models pH neutralization is essential for NADPH oxidase action [51]. DPI inhibited NADPH oxidase and the activity of NHE-1 [52]. NADPH oxidase activity could be different in obese subjects since obese animal models exhibit increased NADPH oxidase-production of superoxide [53]. Furthermore, in monocytes obtained from obese subjects a prolonged production of reactive oxygen species was reported [54].
The differences we observed between lean and obese subjects suggest that obese subjects could have signaling defects in NHE-1 activation pathways. Alternatively, these differences could reflect the small sample size and the fact that among the obese group some individuals were insulin sensitive while others were insulin resistant. This is a limitation of our study but our findings allow power calculations for future studies.
In previous studies we found that rosiglitazone inhibited some NHE-1-mediated actions in monocytes. It was suggested that rosiglitazone could have an ion-transport action [55] and act on NHE-1 [13]. Furthermore, there is a peroxisome proliferator-activated receptor γ (PPARγ) element in the promoter region of NHE-1 [56] indicating a possible interaction between them. It would also be of interest in future studies to investigate whether rosiglitazone can directly influence NHE-1 activity.
It would be of interest to assess the effect of other mediators that are abnormal in obesity (e.g. adiponectin, resistin, ghrelin and visfatin) [57, 58] on NHE-1 activity.
Glucose, insulin, leptin and adrenaline may be increased in obese subjects [59, 60] and monocytes are involved in atherogenesis [61]. Inhibition of the action of these mediators on NHE-1 in lean and obese subjects may be beneficial in the prevention and treatment of atherogenesis. NHE-1 and the signaling molecules involved in its activation are potential therapeutic targets in obesity.
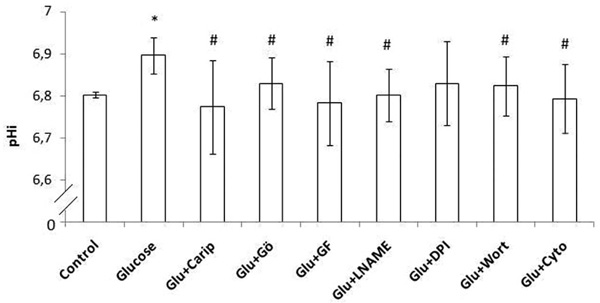
Intracellular pH (pHi) in human monocytes. Glucose was added and pHi was estimated. Monocytes were pre-incubated with cariporide or one of the inhibitors (Gö6976 inhibits α, β and γ isoforms of PKC, GF109203X inhibits all isoforms of the PKC, L-NAME inhibits NOS, DPI inhibits NADPH oxidase, wortmannin inhibits PI3K, cytochalasin D inhibits actin polymerization) and then glucose was added. Error bars indicate standard deviation (SD).
* p < 0.05 vs the respective baseline sample (control sample)
# p < 0.05 vs the respective glucose sample
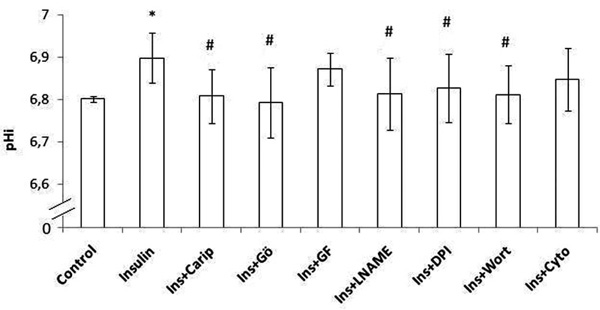
Intracellular pH (pHi) in human monocytes. Insulin was added and pHi was estimated. Monocytes were pre-incubated with cariporide or one of the inhibitors (Gö6976 inhibits α, β and γ isoforms of PKC, GF109203X inhibits all isoforms of the PKC, L-NAME inhibits NOS, DPI inhibits NADPH oxidase, wortmannin inhibits PI3K, cytochalasin D inhibits actin polymerization) and then insulin was added. Error bars indicate standard deviation (SD).
* p < 0.05 vs the respective baseline sample (control sample)
# p < 0.05 vs the respective insulin sample

Intracellular pH (pHi) in human monocytes. Leptin was added and pHi was estimated. Monocytes were pre-incubated with cariporide or one of the inhibitors (Gö6976 inhibits α, β and γ isoforms of PKC, GF109203X inhibits all isoforms of the PKC, L-NAME inhibits NOS, DPI inhibits NADPH oxidase, wortmannin inhibits PI3K, cytochalasin D inhibits actin polymerization) and then leptin was added. Error bars indicate standard deviation (SD).
* p < 0.05 vs the respective baseline sample (control sample)
# p < 0.05 vs the respective leptin sample
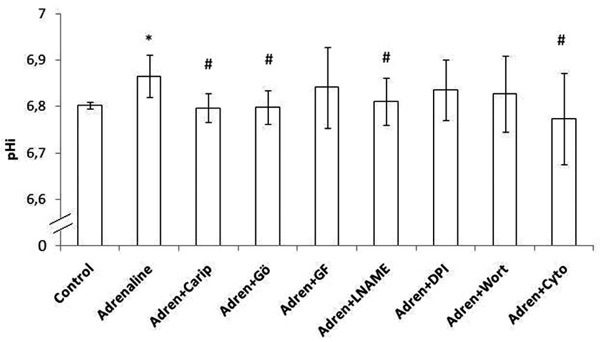
Intracellular pH (pHi) in human monocytes. Adrenaline was added and pHi was estimated. Monocytes were pre-incubated with cariporide or one of the inhibitors (Gö6976 inhibits α, β and γ isoforms of PKC, GF109203X inhibits all isoforms of the PKC, L-NAME inhibits NOS, DPI inhibits NADPH oxidase, wortmannin inhibits PI3K, cytochalasin D inhibits actin polymerization) and then adrenaline was added. Error bars indicate standard deviation (SD).
* p < 0.05 vs the respective baseline sample (control sample)
# p < 0.05 vs the respective adrenaline sample
ACKNOWLEDGEMENTS
M.S. is supported by a grant awarded by the Hellenic Atherosclerosis Society. A.T. is a Senior Research Fellow of the Tseu Medical Institute within Harris Manchester College, Oxford, United Kingdom, and acknowledges funding support.

