All published articles of this journal are available on ScienceDirect.

Cell Membrane Modification for Rapid Display of Bi-Functional Peptides: A Novel Approach to Reduce Complement Activation
Authors Info & Affiliations
Abstract
Ischemia and reperfusion of organs is an unavoidable consequence of transplantation. Inflammatory events associated with reperfusion injury are in part attributed to excessive complement activation. Systemic administration of complement inhibitors reduces reperfusion injury but leaves patients vulnerable to infection. Here, we report a novel therapeutic strategy that decorates cells with an anti-complement peptide. An analog of the C3 convertase inhibitor Compstatin (C) was synthesized with a hexahistidine (His6) tag to create C-His6. To decorate cell membranes with C-His6, fusogenic lipid vesicles (FLVs) were used to incorporate lipids with nickel (Ni2+) tethers into cell membranes, and these could then couple with C-His6. Ni2+ tether levels to display C-His6 were modulated by changing FLV formulation, FLV incubation time and FLV levels. SKOV-3 cells decorated with C-His6 effectively reduced complement deposition in a classical complement activation assay. We conclude that our therapeutic approach appears promising for local ex vivo treatment of transplanted organs to reduce complement-mediated reperfusion injury.
INTRODUCTION
Since the original description of complement activation observed in ischemic hearts [1], there is growing evidence that complement is a central mediator of ischemia/reperfusion (IR) injury (IRI) [2-4]. After decades of neglect, complement has been rediscovered as a potent mediator of inflammation and rejection in organ transplants. Studies have shown that IR-induced complement activation leads to the formation of several key inflammatory mediators that alter vascular homeostasis and stimulate leukocyte activation and chemotaxis [2, 4-6]. Depending on the type of tissue involved, IR-induced complement activation may occur via the classical and/or alternative pathways [4, 7]. Under normal conditions, the vascular endothelial cells express CD59, CD46, CD55 and C receptor 1 (CR1), which offer protection against constant low-level complement activation in plasma [8]. However, following organ transplantation, the inevitable IR activates complement to greater levels than normal, rapidly overwhelming natural anti-complement defenses. To curtail IR-induced or unwanted complement activation, soluble forms of CR1, CD59, CD46 and CD55 have been developed [9, 10], but due to their non-specific nature these agents inactivate complement systemically thereby placing the recipients at increased risk of iatrogenic disease.
At present, second generation agents aimed at targeting specific cells or tissues are being developed for clinical use [11-15]. However, the success rate has been disappointingly low. Some of these targeted agents continue to have drawbacks, including: 1) Systemic delivery with generalized complement suppression; 2) Dosing issues due to dependence on expression levels of targeted membrane proteins, which varies between individuals and/or in the presence of pathological conditions; and 3) Cost effectiveness of recombinant protein products requiring multiple manufacturing steps, which may prove prohibitive for clinical use [11]. To address these issues, we developed a novel and relatively inexpensive therapeutic approach for the local delivery of synthetic anti-complement peptides to be used in transplantation or in by-pass procedures. Our approach takes advantage of an inherent property of small unilamellar fusogenic lipid vesicles (FLVs) [16], which when fusing with cells incorporate their lipids into cell membranes. FLVs are formulated with a mixture of three lipids, one of them containing a nickel (Ni2+) tether. After FLVs fuse with cells, Ni2+ tethers are displayed on the membrane surface and can be used as linkers to decorate cells with a bi-functional peptide comprised of a hexahistidine (His6) Ni2+-tether-binding domain and an anti-complement domain. Liposomes formulated with lipids containing functional groups to bind proteins or antibodies for either targeting or to increase fusogenicity have been previously reported (reviewed in reference [17]). Liposomes formulated with a metal chelating lipid have been incubated with tumor cells with the purpose of displaying recombinant co-stimulatory proteins for potential use as anticancer vaccines [18]. To our knowledge, the use of FLVs to display anti-complement peptides on the surface of cell membranes to control complement activation has not been reported previously.
The purpose of this study was to determine whether a therapeutic strategy based on the decoration of cells with a small bi-functional anti-complement peptide would be effective in reducing complement deposition in vitro. Compstatin (C), a small peptide known to inhibit classical or alternative C3 convertase by preventing C3 cleavage [19-22], was selected to create a bi-functional analog by adding a His6-tag to C and produce C-His6. Here, we report that cells treated with FLVs containing Ni2+ tethers and incubated with C-His6, displayed the bi-functional peptide on cell membranes and effectively reduced complement deposition in a classical activation assay.
MATERIALS AND METHODS
C-His6 Synthesis
The original C peptide sequence Ile1-[Cys2-Val3-Val4-Gln5-Asp6-Trp7-Gly8-His9-His10-Arg11-Cys12]-Thr13-NH2 was modified as previously described to increase potency with substitutions: Val4 to 2-naphthylalanine (2Nal) and His9 to Ala [23]. Furthermore, a His6-tag was added to the C-terminus of the peptide. Solid phase peptide synthesis using Fmoc strategy on a Rink amide resin was used to synthesize C-His6. The peptide was purified using HPLC.
C-His6 Bioactivity Assay
Antibody-sensitized sheep erythrocytes (Sigma-Aldrich, St. Louis, MO) were used to perform a hemolytic complement assay to test C-His6 inhibition. The cells were washed and re-suspended in Gelatin Veronal Buffer (GVB) (Sigma-Aldrich, St. Louis, MO) at a concentration of 1x108 cells/mL and then aliquoted in a 96 well microtiter plate at 100μL/well. C-His6 was serially diluted in 1:20 human serum/GVB from 0.015µM to 0.0039µM in 2:3 parts/volume ratio. 100μL of this solution was then added to the cells and incubated with constant shaking for an hour at 37°C. 150μL of ice-cold GVB was added to all the wells to inhibit further complement activity. The plate was then centrifuged at 800G for 10 min at 4°C. After removing the supernatants, they were transferred to a 96 well microtiter plate and absorbance readings were obtained at 414nm. The 100% lysis value was obtained by suspending cells in water and the minimal value was obtained by suspending cells in GVB only.
Cell Culture
Human skin fibroblasts were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (Sigma Inc. St Louis, MO) supplemented with: 10% Fetal Bovine Serum (FBS), 2mM L-Glutamine and 100U/mL of Penicillin/Streptomycin mixture. Ovarian carcinoma (SKOV-3) cells were incubated in McCoy’s Complete Medium (Caisson Labs, North Logan, UT) with 10% FBS, 2mM L-Glutamine and 100U/mL of Penicillin/Streptomycin. Both cell types were then seeded in 75cm2 flasks and maintained in a tissue culture incubator at 37°C with 5% CO2 and 90% humidity.
Fusogenic Lipid Vesicle Synthesis
FLVs were formulated with various molar ratios of 1,2-Dioleoyl-sn-Glycero-3-Phosphocholine (DOPC), 1-Palmitoyl-2-Oleoyl-sn-Glycero-3-Phosphate (Monosodium Salt) (POPA) and 1,2-Dioleoyl-sn-glycero-3-{[N(5-amino-1-carboxypentyl) iminodiacetic acid] Succinyl} (Nickel Salt) (DOGS-NTA-Ni2+) (Avanti Polar Lipids, Alabaster, AL). The molar ratios of DOPC, POPA, and DOGS-NTA-Ni2+ were varied to optimize Ni2+ tether incorporation onto cell membranes. Lipids in liquid chloroform were mixed in a glass flask, and the mixture was dried under nitrogen gas. The mixture was further desiccated overnight in a vacuum chamber. The lipids were rehydrated in Ca2+/Mg2+-free PBS buffer at a concentration of 1mg/mL, vortexed for 1 min and incubated in a 37°C bath for 5 min. This cycle was repeated 6 times, and the resulting solutions of multi-lamellar lipid vesicles were sonicated with a Branson Sonifier® 450A (Branson Ultrasonics Corp, Danbury, CT) at 50% duty cycle for 1 min/mL to form smaller unilamellar FLVs.
Functionalization of Cell Membranes with Ni2+ Tethers
These experiments were performed using fibroblasts and SKOV-3 cells. FLV formulations were optimized using fibroblasts because of their hardiness and excellent ability to remain attached to well bottoms. Subsequent experiments were conducted on SKOV-3 cells to characterize the effects of FLV levels and incubation time prior to performance of complement deposition studies on these cells. FLV formulations were optimized by varying the molar ratio of DOGS-NTA-Ni2+ thus changing the final levels of Ni2+chelated lipid from 40 to 87ng/µL. Fibroblasts were first seeded in a 96 well microplate coated with 0.5% porcine gelatin solution (Sigma-Aldrich, St. Louis, MO) at a density of 8 x 104 cells/well and incubated for 12 hr at 37°C with 5% CO2 and 90% humidity. Cells were then washed twice with PBS and treated for 20 min with FLVs formulated with the following DOGS-NTA-Ni2+ concentrations: 40, 52, 64, 75 and 87 ng/µL. Eight wells per lipid concentration were tested. To assess Ni2+ tether incorporation, cells were incubated for 30 min with 0.2ng/µL of truncated His6-Cardiotrophin (BioVendor Laboratory Medicine, Inc. Brno, Czech Republic) and with 0.01 μg/μL of FITC- labeled anti-His6 antibody (Alpha Diagnostic, San Antonio, TX) for 15 min. (His6-Cardiotrophin was used as a probe to demonstrate that other His6-tagged agents would couple to lipid-tethered Ni2+ and that the FITC- labeled anti-His6 antibody was an effective method to quantify Ni2+ tethers on membranes). All the wells were washed thrice with PBS between incubation treatments. Background controls did not receive FLVs but were incubated with His6-Cardiotrophin and FITC- labeled anti-His6 antibody. Antibody-specific fluorescence was measured using a SpectraMax M2 (Molecular Devices, Sunnyvale, CA) microplate reader at 494nm excitation and 518nm emission. Reported values were corrected for background.
Similar experiments were performed with SKOV-3 ovarian carcinoma cells to determine the effect of FLV concentration on tether incorporation and C-His6 binding. Cells were seeded at a density of 8 x 104 cells/well in 96 well microplates and exposed for 20 min to FLVs formulated with 64ng/µL DOGS-NTA-Ni2+ diluted in PBS to achieve final lipid concentrations ranging from 12.5 to 500μg/mL. Cells were incubated with 0.01μg/μL of C-His6 for 30 min and with FITC- labeled anti-His6 antibody for 15 min. All wells were washed thrice between steps, and fluorescence was measured as above.
The relation between FLV incubation time and incorporation of Ni2+ tethers was characterized by incubating SKOV-3 cells for 10 to 50 min with FLVs (500µg/mL) formulated with DOGS-NTA-Ni2+ (64ng/µL). Cells were incubated with 0.01μg/μL of C-His6 for 30 min and with FITC- labeled anti-His6 antibody for 15 min. All wells were washed thrice between steps, and fluorescence was measured as described above.
Quantification of C-His6 Levels on Cell Membranes
To quantify C-His6 levels displayed on cell membranes following FLV/C-His6 treatment, a direct analysis assay using UV absorbance was performed on a SpectraMax M2 (Molecular Devices) microplate reader. The assay was based on detecting the 2Nal residue of the C-His6 sequence, which we estimated to have a peak absorbance in the 280-290nm range. To determine the optimal wavelength for C-His6 absorbance, various concentrations of the peptide were serially diluted in half starting at 0.2µg/µL and ending at 0.00625µg/µL. Three 100 µL aliquots per dilution were placed on a 96-well plate. The UV absorbance was measured repeatedly at 2nm increments starting at 270nm until a maximum difference between dilutions was observed, measurements continued at higher wavelengths until differences between samples were no longer apparent at 300nm.
To quantify membrane-bound C-His6 levels, SKOV-3 cells were seeded in Costar 12 well plates and allowed to reach confluence. Cells were incubated for 50 min with FLVs (500µg/mL) and without FLVs. All cells were washed thrice and incubated with 1mL of 0.10 µg/µL of C-His6 for 30 min. Cells were washed thrice, and to release membrane-bound C-His6, cells were incubated with 330µL of PBS (pH 4) for 10 min. 100µL from each well were drawn, placed in a 96 well microplate and UV absorbance was measured at 284 nm. The mean absorbance values were converted to C-His6 levels using a standard curve of known concentrations and adjusted to levels per mL. C-His6 levels per membrane area were calculated using the well’s bottom surface area (38.3mm2) under the assumption that this area was equivalent to the cells’ non-adherent membrane area at 100% confluence.
Time Course Experiments
To determine the duration of C-His6 display on cell membranes, fibroblasts were seeded in Costar 12 well plates and allowed to reach ~90% confluence. Cells were incubated for 30 min with FLVs (500µg/mL) and without FLVs. All cells were washed twice and incubated with 1mL of 0.1 mg/mL of C-His6 for 30 min. Cells were washed and incubated in cell media. Levels of C-His6 were quantified after treatment, at 24 and 48 hours. The same methods used to quantify C-His6 levels displayed on cell membranes in the C-His6 quantification section above were used in these experiments. Membrane-bound C-His6 was released by incubating cells in 330µL of PBS (pH 4) for 10 min. Three 100µL aliquots from each well were drawn, placed in separate wells in a 96 well microplate, and UV absorbance was measured at 284 nm. Absorbance values were converted to C-His6 levels in solution using a standard curve, and levels of peptide at the various time points were compared. The experiment was repeated five times and the results were averaged.
Complement C3b Deposition Assay
We used the protocol described by B.P. Morgan with some modifications to quantify the inhibitory effect of C-His6 [24]. SKOV-3 cells were seeded in 96 well microtiter plates and allowed to reach 90% confluence. Cell membranes were functionalized with Ni2+ tethers by incubating for 40 min with FLVs containing 64ng/μL of DOGS-NTA-Ni2+. Cells were subsequently incubated for 30 min with various concentrations of C-His6 (0.05-0.2 μg/μL). Control cells did not receive any treatment. Cells were then lifted off the plate with CellstripperTM (Mediatech Inc Herndon, VA), washed in 3% BSA/PBS and re-suspended at 1x106 cells/mL in flow cytometry tubes. Trastuzumab (Herceptin) was then added at 0.05μg/μL and the cells were kept at 4°C for 30 min. Each tube was washed with 2mL of McCoy’s medium, centrifuged at 300G for 10 min and re-suspended in 1:2 dilution of fresh human serum. The samples were subsequently incubated at 37°C, 5% CO2, 90% humidity for 30 min with intermittent agitation, washed again in minimal media (M119, 1%FBS) and re-suspended in 1:100 dilution FITC-conjugated goat anti-human F(ab’)2 fragment antibody to C3 (MP-Biomedicals, Solomon, OH). The cells were kept at 4°C for 30 min then centrifuged for 5 min at 300G at 4°C, and re-suspended in 500μL of minimal media. 10μL of propidium iodide (PI) was added to identify dead cells. All cells were washed with PBS between treatment steps. Control groups without C-His6 were included to assess spontaneous complement deposition, non-specific antibody binding, and maximal complement deposition. PI-positive cells were excluded from the analyzed sample and fluorescence emitted by live cells in the FITC channel was quantified for control and experimental groups using the FlowJo software program (Tree Star, Inc., Ashland, OR).
Statistical Analysis
Data from formulation, concentration and incubation time experiments were analyzed for statistical significance by a one-way ANOVA followed by post-hoc pairwise comparisons. Differences between control groups and experimental groups were evaluated using Tukey’s test. In experiments in which two groups were being compared, a student’s t-test was performed. The differences between treatment and control groups as measured by flow cytometry was determined with a chi-square univariate analysis (FlowJo, version 8.8.6) [25]. A P value of <0.05 was considered significant. Data is reported as means ± SEM.
RESULTS
Bioactivity of C-His6
Once C-His6 was synthesized and purified to >95% purity, the peptide was tested for bioactivity using a hemolytic complement assay. The normalized dose response relationship between C-His6 concentration and hemolytic inhibition are shown in Fig. (1). The EC50 was 9.37nM indicating that modification with a His6 tag did not render C-His6 inactive.
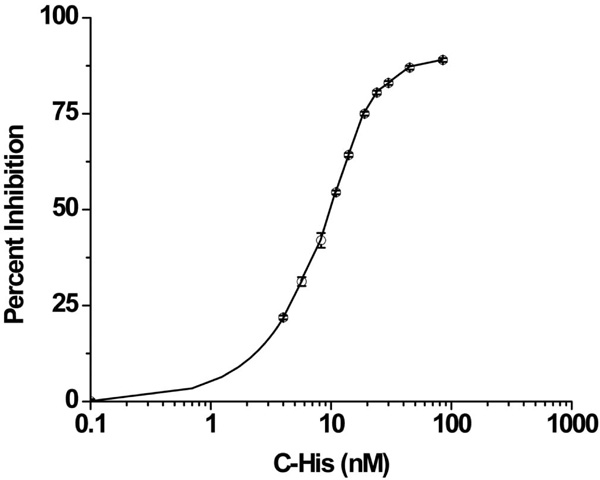
C-His6 inhibition curve of complement mediated erythrocyte lysis. Antibody sensitized sheep erythrocytes were suspended in 1:20 human serum/gelatin veronal buffer (GVB) and treated with serial dilutions of C-His6. The 100% lysis value was obtained by suspending cells in water and the minimal value was obtained by suspending cells in GVB only. The normalized data gave an EC50 value of 9.37nM indicating that the peptide remained bioactive after incorporating the His6-tag.
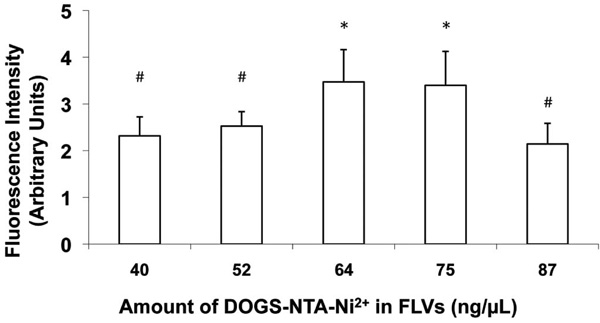
Varying levels of DOGS-NTA-Ni2+ in FLV formulations altered Ni2+ tether incorporation in fibroblast cell membranes. Cells were incubated (20 min) with FLVs formulated with various molar ratios of DOPC : POPA : DOGS-NTA-Ni2+ (n=8 wells per formulation). Cells were washed and incubated (30min) with a truncated His6-tagged protein (Cardiotrophin-His6), an additional wash was performed, and cells were incubated (15 min) with FITC-labeled anti-His6 antibodies. FLVs formulated with 64 and 75ng/µL of DOGS-NTA-Ni2+ bound greater amounts of Cardiotrophin-His6, indicating greater incorporation of Ni2+ tethers. Bars represent mean fluorescence levels ± SEM. * indicates P< 0.05 compared to #.
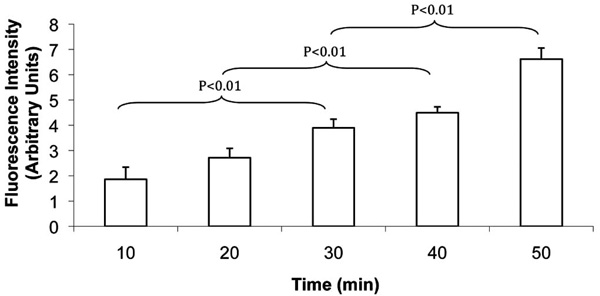
FLV incubation incorporated Ni2+ tethers in cell membranes in a time-dependent manner. SKOV-3 cells were incubated with FLVs for various time periods (n=8 wells per time period). Cells were washed and incubated (30 min) with C-His6, an additional wash was performed, and cells were incubated (15 min) with FITC-labeled anti-His6 antibodies. Bars represent mean fluorescence levels ± SEM.
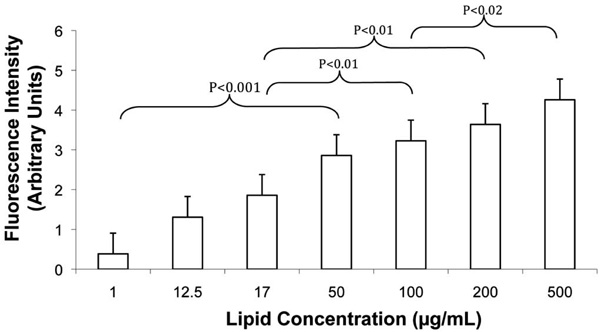
Increasing FLV levels increased Ni2+ tether incorporation in SKOV-3 cell membranes in a dose-dependent manner. Cells were incubated (20 min) with various concentrations of FLVs (n=8 wells per lipid concentration). Cells were washed and incubated (30 min) with C-His6, an additional wash was performed, and cells were incubated (15 min) with FITC-labeled anti-His6 antibodies. Bars represent mean fluorescence levels ± SEM.
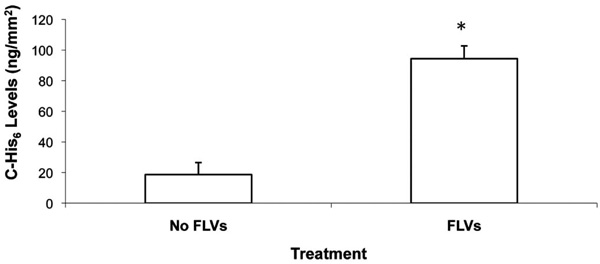
Levels of Ni2+-tether-bound C-His6 per area of cell membrane is shown. Cells (100% confluence) in 12-well plates were incubated (50 min) with and without FLVs. Cells were washed and incubated (30 min) with 1mL of 0.10 mg/mL of C-His6. Cells were washed, and membrane-bound C-His6 was released by incubating cells for 10 min with 330µL of PBS (pH 4). To quantify C-His6, three 100µL aliquots from each well were drawn, placed in separate wells of a 96-well plate and UV absorbance was measured at 284 nm. C-His6 levels per surface area were calculated using a C-His6 standard curve and the surface area of the well’s bottom (38.3mm2). * indicates P< 0.05 when compared to cells without FLV treatment.
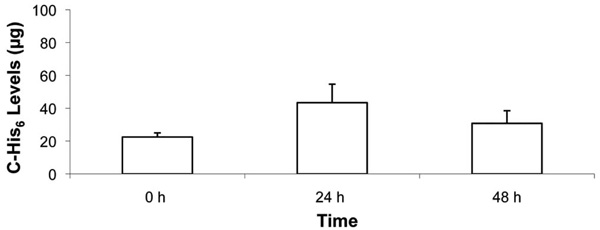
Time course of C-His6 displayed on fibroblast membranes following FLV/C-His6 treatment. Cells in 12-well plates were allowed to reach ~90% confluence. Cells were incubated (30 min) with and without FLVs. Cells were washed and incubated (30 min) with 1mL of 0.1 mg/mL of C-His6. Cells were washed and incubated at 37°C in cell media for 0, 24 and 48 h. At the end of each time point, cells were washed, and membrane-bound C-His6 was released by incubating cells (10 min) with 330µL of PBS (pH 4). To quantify C-His6, three 100µL aliquots from each well were drawn, placed in separate wells of a 96-well plate and UV absorbance was measured at 284 nm. C-His6 levels were calculated using a C-His6 standard curve. Five experiments were combined and there was no statistical significance between the three time points.
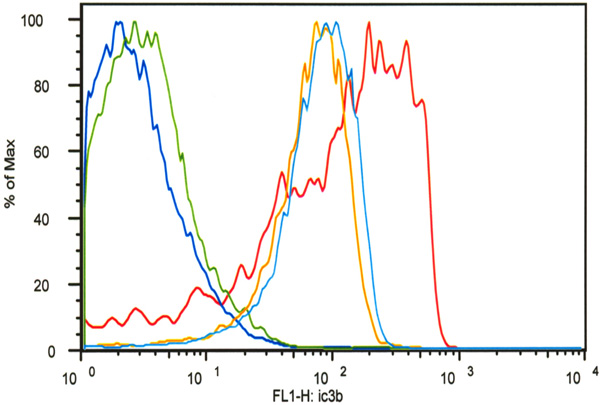
Flow cytometry analysis demonstrated that decoration of SKOV-3 cells with C-His6 reduced complement deposition (C3b) in a classical activation assay. All cell groups except for the untreated controls (dark blue) were incubated with trastuzumab and FITC-labeled anti-C3b antibodies. One group was incubated without serum (green) to determine non-specific C3b antibody binding. One group served as the positive control (red) and two groups were treated with FLVs and C-His6 at two different concentrations: 0.05 mg/mL (light blue) and 0.2mg/mL (orange). FLVs/C-His6 (0.2mg/mL and 0.05mg/mL) treatment reduced C3b deposition significantly compared to untreated positive controls with P< 0.01.
Optimization of FLV Formulation to Maximize Ni2+ Tether Display
To determine composition of FLV formulations that would optimally incorporate Ni+ tethers in membranes, a series of experiments altering levels of DOGS-NTA-Ni2+ were performed. In previous studies (data not shown) which characterized the fusogenicity of FLVs formulated with DOPC-POPA mixtures, we had established that POPA levels are critical to the fusogenic properties of FLVs. In this study, we kept the levels of POPA constant while varying the levels of DOPC and DOGS-NTA-Ni2+. Fig. (2) shows that, as the levels of DOGS-NTA-Ni2+ were increased, Ni2+ tether incorporation appeared to peak significantly at concentrations between 64 and 75ng/µL.
Functionalization of Cell Membranes with Ni2+ Tethers
To further characterize the efficiency of FLVs in incorporating Ni2+ tethers in cell membranes, incubation time and dose response studies were performed. FLVs formulated with DOPC : POPA : DOGS-NTA-Ni2+ at molar percentages of 20:57:23 successfully incorporated Ni2+ tethers on SKOV-3 cell membranes in a time-dependent manner (Fig. 3) and dose-dependent manner (Fig. 4). These results suggest that Ni2+ tether incorporation in membranes can be modulated and carefully controlled by altering FLV incubation time, FLV concentration or both.
Quantification of C-His6 Levels on Cell Membranes
To directly quantify anti-complement peptide levels coupled to Ni2+ tethers on the membrane surface, we performed preliminary studies using UV absorbance, and determined that a wavelength of 284nm was optimal to discern C-His6 levels diluted in PBS. Following FLVs/C-His6 (0.1µg/µL) treatment, the mean levels of three experiments showed that membrane-bound C-His6 was significantly higher (94.4 ± 8.4µg) than in cells treated with C-His6 alone (18.6 ± 7.9µg, P=0.0026). These results indicate that C-His6 was not only binding to Ni+ tethers incorporated in membranes but also binding non-specifically. Fig. (5) shows the calculated levels of C-His6 displayed per surface area of membrane.
Time Course Experiments
To determine whether membrane-localized anti-complement peptide would be displayed by cells for 48 h, fibroblasts were treated with FLVs/C-His6 therapy, and levels of C-His6 were quantified at 0 h, 24 h and 48 h post-treatment. As shown in Fig. (6), C-His6 display was stable over 48 h. There was no statistical significance between mean C-His6 levels released by the low pH buffer at the three time points. However, in all experiments there was a trend for higher C-His6 levels released at 24 h.
Complement C3b Deposition Assay
To determine whether decoration of anti-complement peptide on cell surfaces would decrease complement activation efficiency, we used HER2/neu-overexpressing human ovarian carcinoma SKOV-3 cells. As shown in Fig. (7), anti-HER2/neu antibody could bind to HER2/neu and potently activate complement leading to iC3b deposition (red line). The mean fluorescence intensity (MFI) as measured by flow cytometry of live SKOV-3 cells with anti-HER2/neu antibody plus fresh human serum as a source of complement was 189 ± 171. In contrast, MFI from SKOV-3 cells treated with FLVs and C-His6 was significantly lower than that of untreated control cells, reflecting reduced iC3b deposition. Cells treated with the highest C-His6 concentration, 0.2mg/mL (orange line) had a MFI value of 79.1 ± 43.9 as compared to untreated control (red line) which had a MFI value of 189 ± 171 with P<0.01 for both groups (0.05mg/mL and 0.2 mg/mL). These data suggest that indeed localization of anti-complement peptide to cell surfaces significantly impairs potent complement activation.
DISCUSSION
We present a novel approach of incorporating small peptides onto the surface of cell membranes; an approach that has potential application in the development of anti-complement therapies. The purpose of this study was two-fold. First, we wished to demonstrate that FLVs formulated with a novel combination of lipids can efficiently allow the display of a small anti-complement peptide on cell membranes for at least 48 h. Second, we wanted to provide proof-of-concept that the membrane-immobilized peptide is able to reduce complement deposition in a classical activation assay. Our data provides evidence that the lipids used in our FLV formulation produced vesicles that rapidly fused with cells without the aid of fusogens, and after FLV treatment, cell membranes were functionalized with C-His6, protecting cells from complement activation.
In the literature, various approaches have been reported to modify cell membranes with proteins in vitro [26-29]. In some approaches, proteins were derivatized to permit their direct anchorage to cell membranes by adding lipophilic moieties to proteins such as: glycosyl phosphatidylinositol [26], palmitic acid [28], and hydrophobic tails [27]. Another approach involved the biotinylation of endogenous membrane proteins with sulfo-NHS-LC-biotin to anchor streptavidin-containing chimeric proteins [29]. Although all approaches were effective in decorating cells, there were differences in the time course of display. However, short-term or long-term protein display needs to be considered in the context of the therapeutic agent’s activity requirements.
Liposomes have not been used extensively to modify cell membranes although this potential clearly exists. Liposomes can be shaped and formulated to work either as long-acting sustained release vesicles (low fusogenicity) for drug delivery or as fusogenic vesicles for intracellular delivery of various agents [16, 17, 30-37]. When fusogenic vesicles merge with cells, their lipids incorporate into plasma membranes and provide an opportunity to modify the cell surface. Two research teams have reported modification of cell membranes using fusogenic vesicles. Fadok et al. performed an in vitro study in which T cells treated with fusogenic liposomes formulated with phosphatidylserine induced co-cultured macrophages to engulf what appeared to be apoptotic T cells [38]. van Broekhoven et al., in the process of developing a cancer vaccine, formulated liposomes with α-palmitoyl-β-oleoyl-phosphatidylcholine (POPC) and a zinc-chelating lipid, nitrilotriacetic acid di-tetradecylamine (NTA-DTDA), to display His6-tagged recombinant co-stimulatory molecules on murine P815 tumor cells [18]. Using this technique, mice immunized with P815 tumor cells bearing B7.1 and CD40 demonstrated an increased cytolytic T-lymphocyte activity against P815 cells and inhibited tumor growth. However, their approach had some shortcomings. The vesicle’s lipid formulation and construction was not optimal for promoting fusion with cells and required the aid of a fusogen (i.e., polyethylene glycol). The time course of protein display was relatively short, protein levels (measured using fluorescence intensity) decreased rapidly from ~400-fold (immediately after treatment) to ~60-fold and ~15-fold by 4 and 24 h, respectively. The authors attributed the loss of displayed protein to a predominant internalization of the NTA-DTDA-protein complex by the cell [18]. In the present study, the levels of C-His6 displayed on the membrane surface did not change significantly over 48 h. These results are very different from those obtained by van Broekhoven et al., and we attribute the longer duration of peptide display in our study to important differences in lipid vesicle formulation, which promote vesicle-to-cell fusion without the use of fusogens like polyethylene glycol (see comments below). Once fusion occurred and vesicle lipids incorporated in the cell membrane, we did not observe changes that would suggest internalization of the DOGS-NTA-Ni2+-peptide complex. We observed the opposite, at 24 h there was a trend of increasing C-His6 levels being released from DOGS-NTA-Ni2+. This non-significant trend (P=0.0825) may represent release of either non-specific bound C-His6 or internalized peptide, which interacted and coupled with empty Ni+ tethers over the first 24 h period, and later released by the low pH PBS incubation, producing the apparent trend.
The construction and formulation of FLVs used in the present study differed from those presented by van Broekhoven et al. [18]. Our FLV formulation consisted of DOPC, POPA and DOGS-NTA-Ni2+, which was different from their formulation (POPC and NTA-DTDA). To increase FLV instability and enhance fusion with cells, we added an asymmetric lipid (POPA) to disrupt homogeneity of the lipid bilayer. Furthermore, our FLVs were created with a high-frequency sonicator, to produce vesicle populations with a tight radius of curvature (~125nm), which increases instability of FLV membranes and enhances fusion with cells. Our approach did not require any fusogens to boost incorporation of DOGS-NTA-Ni2+ into the cell membrane. Another important difference between the two approaches is that Ni2+ was chelated to the lipid before FLV construction eliminating the need to add ionized divalent metal to the incubation buffer (a potential source for toxicity) during vesicle-cell fusion. Finally, the therapeutic goal of our approach was not vaccine development, but rather to reduce IRI by reinforcing endogenous complement control proteins expressed on cell membranes with an anti-complement peptide.
The ability to locally deliver complement inhibitors in an efficient manner to sites of inflammation can have tremendous benefits in complement-mediated pathology. One obvious application is in tissue transplantation, which by definition has to undergo IR. In most tissues prolonged IR is known to cause endothelial cell dysfunction due to abnormal increases in the reactive oxygen species, increased expression of adhesion molecules and complement activation [1, 39-43]. All these factors can contribute toward the functional impairment of the endothelial barrier, thereby leading to protein extravasation and interstitial edema. Several studies have also shown that complement activation induced by IR leads to the formation of key inflammatory mediators (C3a, C5a, C3b and C5b-9) that alter vascular homeostasis and stimulate leukocyte activation and chemotaxis [44]. Following transplantation pathological changes observed in the grafts have been linked to increased complement activation. Normally endothelial cells express complement regulatory proteins to self-protect from continuous low-level complement activity in plasma [8]. However, in transplanted grafts following IR, these defenses are rapidly overwhelmed and endothelial cell damage and dysfunction ensues. Our therapeutic approach can enhance endogenous complement control proteins and reduce tissue injury following IR.
Although sCR1 and anti-C5 monoclonal antibody have been successful in offering protection against IRI [45, 46], long-term systemic inhibition of complement can lead to infections and auto-immune diseases [47, 48]. Alternative targeted approaches are being developed, including membrane targeting strategies [8, 9, 13]. sCR1sLex (TP20) consisting of soluble CR1 decorated with fucosylated tetrasaccharide sialyl Lewisx, targets sLex binding sites on E-selectin and P-selectin adhesion molecules expressed on the endothelium after IR. sCR1-3-MSWP1 (APT070), CD59-MSWP1 and DAF-MSWP1 consist of fragments of natural complement control proteins coupled to the membrane targeting peptide 1 (MSWP1). In animal models of transplantation and IR injury, TP20 and APT070 have been shown to be effective [11, 14, 49, 50]. Although cell membrane-targeted agents appear to be promising in IRI animal studies, none have been approved for this indication by the FDA [15].
CONCLUSION
In summary, in this study we selected and modified an anti-complement peptide to work with our FLV delivery system. FLVs allowed control of displayed peptide levels by varying formulation, incubation time, and/or concentration of FLVs. The peptide remained anchored to the site of complement activation for at least 2 days. Based on the results, we conclude that our FLV approach to deliver small peptides has potential for local treatment of tissues to reduce IR-induced complement activation. Our approach appears to be well suited for ex vivo treatment of organs in transplantation. However, to assess efficacy in vivo, future studies will need to be performed in primates since C only inhibits primate-derived complement.
ABBREVIATIONS
CONFLICT OF INTEREST
The authors acknowledge that there is a financial conflict of interest.
ACKNOWLEDGEMENTS
This work was funded in part by grants from the National Institutes of Health R41 HL079855 and from EndoProtech, Inc. Louisville, KY.

