All published articles of this journal are available on ScienceDirect.

ATL 313, A Selective A2A Adenosine Receptor Agonist, Reduces Myocardial Infarct Size in a Rat Ischemia/Reperfusion Model
Abstract
Objective:
The cardioprotective effects of activation of the A2A adenosine receptor (A2AAR) on ischemia/reperfusion injury in the heart remain controversial. We investigated whether ATL 313, a new selective A2AAR agonist, could reduce myocardial infarct size in a rat ischemia/reperfusion model.
Methods:
Sprague-Dawley rats were subjected to a 40 minute occlusion of the left coronary artery followed by 3 hours reperfusion. Hemodynamics were monitored during the procedure. The rats were divided into 3 groups: Group 1 received continuous intravenous infusion of saline given 10 min prior to ischemia and throughout reperfusion (n=8); Group 2 received continuous intravenous infusion of 10 ng/kg/min of ATL 313 given 10 min prior to ischemia, and throughout reperfusion (n=8); and group 3 received an intravenous bolus of ATL 313 (900 ng/Kg body weight) given 10 min prior to ischemia, and continuous intravenous infusion of 10 ng/kg/min of ATL 313 started at 20 min after ischemia and throughout reperfusion (n=8). After euthanasia of the rats, the hearts were harvested for the assessment of risk zone and zone of necrosis of the left ventricle.
Results:
The percentage of risk zone in the left ventricle was similar among group 1 (47 ± 3.7 %), group 2 (41.5 ± 4.2 %) and group 3 (42.4 ± 3.8 %). However, the infarct size, expressed as a percentage of the risk zone, was significantly decreased in group 3 (30.6 ± 5 %, P=0.01) compared with group 1 (53.8 ± 6.2 %) and group 2 (52.1 ± 4.8 %). In group 3, the bolus injection of ATL 313 caused a reduction in blood pressure during the procedure, and decreased heart rate and LV ±dp/dt before coronary artery occlusion; but increased LV +dp/dt at the end of reperfusion compared to the other 2 groups.
Conclusion:
A2AAR agonist ATL313 significantly reduced infarct size and improved LV contractility at the end of reperfusion assessed by LV dp/dt at a dose of 900 ng/Kg. The mechanisms for the observed cardioprotection effect of ATL313 remain to be determined.
Introduction
In the recent AMISTAD II post hoc analysis [1], patients receiving adenosine plus early coronary artery reperfusion had beneficial effects from the treatment including reduced death and smaller infarcts. Activation of adenosine receptors has been shown to reduce myocardial infarct size in experimental models. Hale et al. [2] demonstrated that stimulation of adenosine receptors with an adenosine A1 receptor agonist, R(-)-N-(2-phenylisopropyl)-adenosine (PIA), resulted in a significant reduction in myocardial infarct size in anaesthetised rabbits that were subjected to 40 minutes of coronary artery occlusion followed by three hours of reperfusion. Adenosine can signal through any of four types of G protein-coupled adenosine receptors (A1, A2A, A2B, and A3) [3]. Whether activation of the A2A adenosine receptor (A2AAR) could reduce ischemia/reperfusion injury in the heart remains controversial. In one study, Lasley et al. [4] demonstrated that intracoronary infusions of the A2AAR agonist CGS-21680 reduced myocardial infarct size in open-chest pigs that were subjected to 60 minutes of coronary artery occlusion and 3 hours of reperfusion. In another study, Glover et al. [5] reported that intravenous infusion of the A2AAR agonist ATL-146e reduced myocardial infarct size in a canine model that was subjected to 90 minutes of coronary artery occlusion followed by 2 hours of reperfusion. However, some other studies have shown that A2AAR agonist has no cardioprotective effects when administered prior to the onset of ischemia [6, 7].
A new selective A2AAR agonist 4-{3-[6-amino-9-(5-cyclopropylcarbamoyl-3,4-dihydroxytetrahydrofuran-2-yl)-9H-purin-2-yl]prop- 2-ynyl}piperidine-1-carboxylic acid methyl ester (ATL 313) has been developed. ATL 313 is not metabolized in animals and has a longer half-life (30 minutes) compared to another A2AAR agonist, ATL-146e. Several studies have shown that ATL 313 attenuates ischemia/reperfusion injury in the lung [8, 9]; however, the effect of ATL 313 on myocardial reperfusion injury has not been determined. This study was designed to investigate whether ATL 313 could reduce myocardial infarct size and favorably affect left ventricular (LV) function in a rat ischemia/reperfusion model.
Methods
All experimental protocols were approved by the Institutional Animal Care and Use Committee, and performed in accordance with the “Guide for the Care and Use of Laboratory Animals” (NIH publication No. 85-23, National Academy Press, Washington DC, revised 1996). The Heart Institute at Good Samaritan Hospital is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
ATL 313 preparation
ATL 313 was made as concentrated stock solutions in DMSO at a concentration of 100 µg/ml, and then diluted in phosphate-buffered saline (PBS; pH 7.4) to a concentration of 250 ng/ml.
Animal surgical preparation
Female Sprague-Dawley (SD) rats (190~240g body weight) were weighed and anesthetized with an intraperitoneal administration of ketamine (7.5 mg/100 g body weight) and xylazine (0.5 mg/100 g body weight). Additional anesthesia was given throughout the study as required. The chest, neck and right femoral areas were shaved. The rats were intubated and ventilated with room air (rate 60 cycles/min, tidal volume 1 ml per 100 g body weight), and placed on a heating pad (37 °C). The right carotid artery was exposed, and a 2F high-fidelity, catheter-tipped micromanometer (model SPR-869, Millar, Inc) was advanced into the ascending aorta and left ventricle (LV) to record LV hemodynamic parameters. The jugular vein was canulated to inject ATL 313 or vehicle, and to administer blue dye and potassium chloride at the end of the experiment. The femoral artery was isolated and catheterized to continuously monitor heart rate and arterial blood pressure using an ADI (Advanced Digital Instruments, Grand Junction, CO) system. The chest was opened through the 4th intercostal space. The pericardium was removed. The left coronary artery, which is located in the groove between the pulmonary outflow track and left ventricle, was encircled with 4-0 silk suture at the site just under the left atrium for occluding and reperfusing the artery. All of the rats were subjected to a 40 minutes coronary occlusion followed by 3 hours of reperfusion. At the end of the protocol, the coronary artery was reoccluded and 0.6ml 50% Unisperse blue dye (Ciba Geigy, Hawthorne, NY) was injected intravenously to delineate the risk zone. Animals were euthanized with 1 ml potassium chloride (149 mg/ml I.V.) under deep anesthesia, and the heart was excised.
Experimental groups
Animals were divided into 3 groups. Group 1 received continuous intravenous infusion of PBS given 10 min prior to ischemia and throughout reperfusion (n=8); Group 2 received continuous intravenous infusion of 10 ng/kg/min of ATL 313 (dissolved in PBS) given 10 min prior to ischemia, and throughout reperfusion (n=8); and group 3 received an intravenous bolus of ATL 313 (900 ng/kg body weight; diluted in 0.6 ml PBS) given 10 min prior to ischemia, and continuous intravenous infusion of 10 ng/kg/min of ATL 313 started at 20 min after ischemia and throughout reperfusion (n=8). The infusion rate of the pump was set at a rate of 7.9 µl/min. During the 4 hours and 10 minutes of the procedure, a total volume of 1.98 ml was infused in all animals.
Myocardial Risk Zone and Infarct Size Assessment
The left ventricle was cut into 4 equally thick transverse slices. These were photographed to show the ischemic risk zone (not stained blue, Fig. 1A). The slices were then incubated in 1% triphenyltetrazolium chloride (TTC) for 15 minutes at 37 °C to delineate the necrotic zone (infarcted zone). TTC stains viable myocardial tissue brick red and infarcted tissue appears white (Fig. 1B). The ventricular slices were then rephotographed. Projected photographs of the 3 slices below the coronary artery occlusion site in each heart, showing the risk zone and the necrotic zone, were traced and then digitized using a computerized planimetric system. The areas of ischemic and nonischemic regions were computed and expressed as a percentage of the slice, as were the percentages of necrotic and non-necrotic tissue. The percentage in each slice was then multiplied by the weight of the slice to obtain mass. The masses were summed for each heart. Risk zone (unstained blue zone) was expressed as a percentage of the left ventricular mass. The extent of necrosis was calculated as percentage of the left ventricular mass, and the infarct size expressed as percentage of the ischemic risk zone (infarct size = extent of necrosis/extent of risk zone).
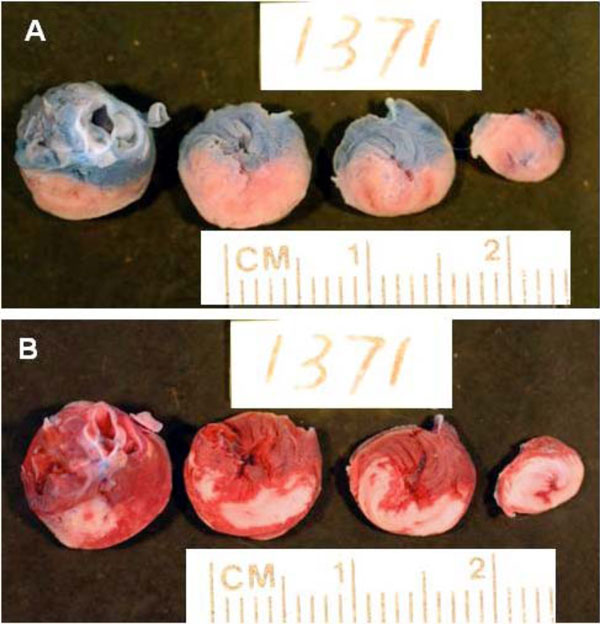
Representative slices of heart from a rat in control group to show risk and necrosis area. A: Slices show the risk area. The area stained by blue dye represents the nonischemic area. The non-blue area (pink) is the ischemic risk zone. B: The same slices as in panel A stained for TTC. Brick red represents viable myocardial tissue and the white area is the necrotic tissue. Note the large homogeneous infarction (white area) of control ischemic myocardium.
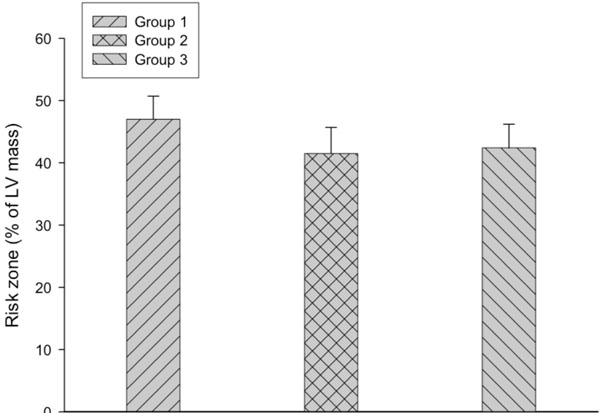
Risk zone is expressed as percentage of LV mass. Risk zones are similar among the 3 groups.
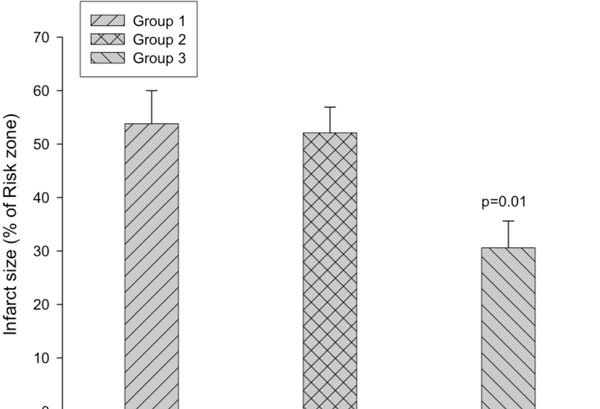
Infarct size is expressed as percentage of the risk zone. The infarct size is significantly decreased in group 3 compared with the other two groups (p=0.01).
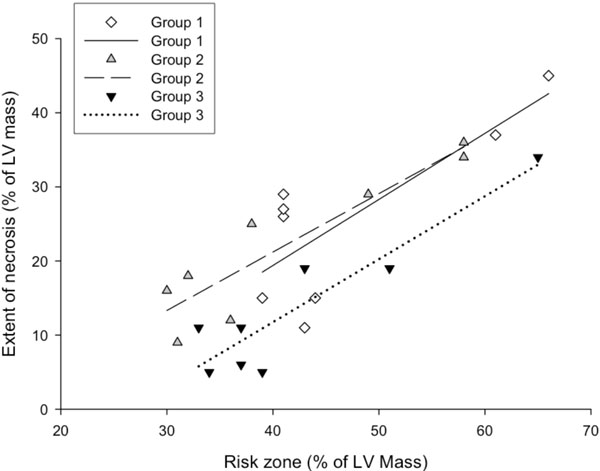
The association between myocardial risk zone (expressed as percentage of LV mass) and extent of necrosis (expressed as percentage of LV mass) in the 3 groups. Note that for any size risk zone, the extent of necrosis was smaller in group 3 compared with the other two groups. P<0.05 (group 3 vs. group 1 and 2) for group effect by ANCOVA.
Statistical Analysis
All data are reported as mean ± SEM. The risk zone and the infarct size, were compared by one-way ANOVA with subsequent Tukey’s post hoc test if appropriate. ANCOVA analysis was used for a group effect on the regression model of risk zone versus the extent of necrosis. Hemodynamic data were analyzed by repeated measures analysis of variance. If an over all significance of f < 0.05 was obtained, data among groups at specific time points were analyzed by ANOVA. Statistically significant differences are established at P<0.05.
Results
Risk Zone, Zone of Necrosis and Infarct Size
The risk zone, which was expressed as the percentage of the left ventricular mass, was similar among group 1 (47 ± 3.7 %), group 2 (41.5 ± 4.2 %) and group 3 (42.4 ± 3.8 %) (Fig. 2). However, infarct size, expressed as a percentage of the risk zone, was significantly decreased in group 3 (30.6 ± 5 %, P=0.01) compared with group 1 (53.8 ± 6.2 %) and group 2 (52.1 ± 4.8 %) (Fig. 3). As shown in Fig. (4), the extent of necrosis, which was expressed as the percentage of the left ventricular mass, was correlated with the risk size in each group (r was 0.92 in group 2 and group 3, and 0.79 in the group 1), and the regression line of the group 3 was shifted downwards. ANCOVA analysis demonstrated that the group effect on the relationship was P<0.05.
Hemodynamics
As shown in Table 1, the injection of the bolus of ATL 313 in group 3 significantly decreased arterial blood pressure during the procedure, suggesting that this dose of ATL 313 caused a vasodilation effect. LV ±dp/dt was significantly lower in group 3 compared to the other 2 groups before coronary artery occlusion. However, ATL 313 in group 3 significantly increased LV +dp/dt at the end of 3 hours of reperfusion. The heart rates were not significantly different among the three groups prior to coronary artery occlusion (Table 1); but at this time point the heart rate was significant lower compared to the baseline in group 3 (p=0.016), suggested ATL 313 decreased heart rate in group 3.
Hemodynamics (n=8 in Each Group)
| Baseline | Group 1 | Group 2 | Group 3 | P value |
|---|---|---|---|---|
| Heart rate (beats/minute) | 242 ± 15 | 241 ± 9 | 235 ± 10 | NS |
| Systolic blood pressure (mmHg) | 118 ± 5 | 117 ± 7 | 115 ± 9 | NS |
| Diastolic blood pressure (mmHg) | 73 ± 5 | 80 ± 6 | 79 ± 5 | NS |
| Mean blood pressure (mmHg) | 88 ± 5 | 92 ± 6 | 91 ± 6 | NS |
| +dp/dt (mmHg/second) | 5281 ± 312 | 5732 ± 246 | 5482 ± 313 | NS |
| -dp/dt (mmHg/second) | 4798 ± 262 | 5126 ± 275 | 4858 ± 303 | NS |
| Prior to coronary artery occlusion | ||||
| Heart rate (beats/minute) | 233 ± 16 | 212 ± 14 | 194 ± 9 | NS |
| Systolic blood pressure (mmHg) | 132 ± 15 | 108 ± 7 | 68 ± 6 | 0.001* |
| Diastolic blood pressure (mmHg) | 76 ± 12 | 73 ± 7 | 35 ± 3 | 0.004* |
| Mean blood pressure (mmHg) | 95 ± 13 | 84 ± 7 | 46 ± 4 | 0.002* |
| +dp/dt (mmHg/second) | 5508 ± 454 | 5274 ± 300 | 3815 ± 376 | 0.01* |
| -dp/dt (mmHg/second) | 4938 ± 468 | 4492 ± 413 | 2660 ± 203 | 0.0008* |
| Prior to coronary artery reperfusion | ||||
| Heart rate (beats/minute) | 221 ± 14 | 204 ± 10 | 203 ± 8 | NS |
| Systolic blood pressure (mmHg) | 82 ± 4 | 77 ± 4 | 69 ± 5 | NS |
| Diastolic blood pressure (mmHg) | 54 ± 4 | 43 ± 3 | 39 ± 3 | 0.012** |
| Mean blood pressure (mmHg) | 64 ± 4 | 54 ± 3 | 49 ± 4 | 0.018** |
| +dp/dt (mmHg/second) | 4069 ± 229 | 3684 ± 162 | 3898 ± 265 | NS |
| -dp/dt (mmHg/second) | 2818 ± 154 | 2639 ± 345 | 2176 ± 120 | NS |
| After 3 hour reperfusion | ||||
| Heart rate (beats/minute) | 210 ± 14 | 211 ± 10 | 231 ± 8 | NS |
| Systolic blood pressure (mmHg) | 78 ± 4 | 70 ± 3 | 71 ± 4 | NS |
| Diastolic blood pressure (mmHg) | 48 ± 4 | 38 ± 2 | 37 ± 2 | 0.027** |
| Mean blood pressure (mmHg) | 58 ± 4 | 49 ± 2 | 48 ± 2 | 0.044** |
| +dp/dt (mmHg/second) | 3358 ± 99 | 3442 ± 182 | 4199 ± 207 | 0.0034* |
| -dp/dt (mmHg/second) | 2385 ± 125 | 2182 ± 154 | 2400 ± 229 | NS |
NS = not significant.
* significant difference (group 3 vs. other 2 groups);
** significance difference (group 3 vs. group 1).
Discussion
The results of the present study indicated that the selective A2AAR agonist, ATL 313, reduced myocardial infarct size and improved LV contractility at a dose with systemic hemodynamic effects in a rat ischemia/reperfusion model. However, this beneficial effect was not present in a dose below the threshold for producing systemic hemodynamic effects.
A number of studies have shown that A2AAR agonists reduce infarct size in ischemic/reperfused myocardium. For example, Jordan et al. [10] demonstrated that intracoronary infusion of the selective A2AAR agonist CGS-21680 (started at 5 minutes before reperfusion and discontinued after 60 min) significantly reduced infarction in anesthetized, open-chest dogs subjected to 60 minutes of left anterior descending coronary artery occlusion and 3 hr reperfusion. Glover et al. [5] observed that intravenous infusion of A2AAR agonist ATL-146e (begun at 5 minute before ischemia and continued throughout the protocol at a low, nonvasodilating dose) reduced infarct size in a canine model that was subjected to 90 minutes of coronary artery occlusion followed by 2 hours of reperfusion. Yang et al. [11] also found that A2AAR activation by ATL 146e reduced infarct size in C57BL/6 mice after 45 minutes of left coronary artery occlusion followed by 24 hours of reperfusion. Smits et al. [12] administered adenosine A1/A2 receptor agonist AMP 579 before ischemia (bolus loading dose i.v. followed by continuous i.v. infusion) in a porcine model of myocardial infarction (40 minute LAD occlusion followed by 3 hour reperfusion). AMP 579 significantly reduced infarct size, and suggested that the cardioprotective effect of AMP 579 was due to stimulation of adenosine A2 receptors. However, not all studies have shown a beneficial effect of A2AAR activation. Thornton et al. [6] reported that pretreatment with a selective A2AAR agonist CGS 21680 (administered at 15 minutes before coronary occlusion) caused hypotension but failed to limit myocardial infarct size in rabbits that were subjected to 30 minutes of regional coronary ischemia and 3 hours of reperfusion. Lasley et al. [7] also demonstrated that preischemic administration of the A2AAR agonist CGS-21680 did not reduce myocardial infarct size in rats submitted to 25 minutes regional myocardial ischemia and 2 h reperfusion. In our present study, A2AAR agonist ATL 313 reduced infarct size in a dose with systemic hemodynamic effects in rat ischemia/reperfusion model. The difference in outcome between the present study and other studies may be due to the different A2AAR agonists utilized, different animal species or experimental protocol (particularly with regard to timing of administration and dosage).
The beneficial effects of treatment with A2AAR agonists may occur on the cardiac myocytes. A2AAR is also distributed in various cardiovascular tissue including atria, ventricle and coronary artery [13]. Activation of A2AAR can increase inotropy of the ventricular myocytes [14] and/or improve left ventricular function. Lasley et al. [4] found that low-dose intracoronary infusions of A2AAR agonist CGS-21680 increased contractility in an in vivo stunned porcine myocardium associated with a small decrease in arterial blood pressure. In contrast, Kilpatrick et al. [15] reported that treatment of isolated rat ventricular myocytes with the A2AAR agonist CGS-21680 failed to alter their contractility, although A2AAR were expressed in myocytes. Kilpatrick’s results are consistent with that of a lack of effect of CGS-21680 in normal intact myocardium [4]. These conflicting results suggested that A2AAR activation may induce different effects on contractility in normal and stunned myocardium. Our present data showed that ATL 313 significantly decreased LV ±dp/dt initially before coronary artery occlusion (normal myocardium) in a dose with systemic hemodynamic effects, but significantly increased LV +dp/dt at the end of 3 hours of reperfusion (injured myocardium), suggesting that ATL 313 improved LV contractility in reperfused myocardium. It is possible that during ischemia the reduction in afterload and contractility of the left ventricle reduced myocardial oxygen demand, which then reduced myocardial infarct size. Upon reperfusion, the reduced infarct size coupled with enhanced contraction of the stunned myocardium may have explained the improved LV +dp/dt at the end of reperfusion.
Activation of A2AAR in cardiovascular tissue also can cause vasorelaxation in coronary arteries [16]. Whether the cardioprotective effects of A2AAR activation are due to this vasorelaxation remains unknown. Glover et al. [5] showed that A2AAR agonist ATL-146e reduced infarct size without causing vasodilatation using very low, nonvasodilating doses in a canine model of reperfused myocardial infarction. Yang et al. [11] also found that ATL 146e reduced infarct size after 45 minutes of left coronary artery occlusion followed by 24 hours of reperfusion at a dose that had no effect on heart rate, peripheral arterial pressure, or LV pressures in B6 mice. These experimental studies all demonstrated that cardioprotection observed with A2AAR agonists is not due to direct effect on the vasculature. However, in our present study using the rat ischemia/reperfusion model, A2AAR agonist ATL 313 did not reduce myocardial infarct size at a dose without vasodilating effects, although it did reduce infarct size at a dose with vasodilating effects.
Our present study demonstrated that the reduction in infarct size observed in group 3 was associated with systemic hemodynamic effects. ATL 313 reduced blood pressure to 40% of the baseline value. While a transient fall in blood pressure prior to coronary artery occlusion may result in a preconditioning like effect [17], the fall in blood pressure with the ATL 313 was not transient, but persistent – hence, it really did not mimic a transient preconditioning-like fall in blood pressure. In addition, several studies have demonstrated that systemic hypotension before coronary artery occlusion did not necessarily induce cardioprotection [6, 7]. Thornton et al. [6] and Lasley et al. [7] reported that pretreatment with a selective A2AAR agonist CGS 21680 before coronary occlusion caused hypotension but failed to limit myocardial infarct size in rabbit and rat ischemia/reperfusion models. These data suggested that a decrease in blood pressure before coronary artery occlusion may not be the sole cause of the beneficial effects of ATL 313 in our study. The systemic hypotension may be overcome by giving ATL 313 into the coronary circulation to achieve a higher cardiac tissue level instead of by intravenous injection. On the other hand a reduction in blood pressure during the ischemic event may benefit the heart by reducing oxygen demand (reduced afterload).
It is known that stimulation of A1AR (A1 adenosine receptor) results in a negative chronotropic and inotropic effect, while pure A2AAR activation inhibits neutrophil function and dilates vascular smooth muscle [6]. Thornton et al. [6] and Lasley et al. [7] have demonstrated the cardioprotection of stimulating A1AR by pretreatment with A1AR agonist before coronary occlusion in rabbit and rat model. Sun et al. [18] reported that the specificity of ATL313 for A2AAR was approximately 100-fold greater selectivity compared to A1AR and A3AR. In our present study, ATL313 induced significant decrease in heart rate and LV dp/dt – suggested the dose of ATL313 was high enough to activate A1AR in group 3. Therefore, the cardioprotection of ATL313 in group 3 may be due to a preconditioning effect by activation of A1AR in a high dose with systemic hemodynamic effects, and not by pure activation of A2AAR.
There are several limitations in our present study. One limitation is that the bolus loading dose of ATL 313 induced profound hypotension in group 3, while hypotension during the acute course of myocardial infarction increases mortality in the clinical setting. Further studies are needed to investigate whether lower does of ATL 313, which can increase intracellular cyclic AMP but did not cause systemic hypotension, has cardioprotective effects. The second limitation is that bolus loading dose before ischemia plus continuous intravenous infusion was used in group 3. It is not clear whether the observed cardioprotective effects are due to increased resistance to ischemia or anti-reperfusion injury. Whether administration of ATL 313 starting before reperfusion, which may be more clinically relevant, has cardioprotective effects are needed. The third limitation is that we did not include a control group, with artificially reduced blood pressure, to allow differentiating cardioprotective effects of the medication versus hemodynamic effects. Whether hypotension alone induced the cardioprotective effects in the present study will require further investigation.
In summary, the A2AAR agonist ATL313 significantly reduced infarct size and improved LV contractility at a dose with systemic hemodynamic effects in a rat myocardial ischemia/reperfusion model. The mechanism of these beneficial effects is not well understood. Whether cardioprotection by ATL313 is due to its actions on the vasculature, or through the preconditioning effect by stimulating A1AR, will require further investigation.
ACKNOWLEDGEMENT
We thank Adenosine Therapeutics for providing the ATL 313.

