All published articles of this journal are available on ScienceDirect.

Vascular Function and Inflammation in Rheumatoid Arthritis: the Role of Physical Activity
Authors Info & Affiliations
Abstract
Inflammation disturbs biochemical pathways involved in homeostasis of the endothelium. Research has established clear links between inflammatory mediators, particularly C-reactive protein and tumour necrosis factor alpha, endothelial dysfunction, and atherosclerosis. Endothelial dysfunction and atherosclerosis may be subclinical at early stages, and thus the ability to detect them with non-invasive techniques is crucially important, particularly in populations at increased risk for cardiovascular disease, such as those with rheumatoid arthritis. This may allow the identification of interventions that may reverse these processes early on. One of the best non-pharmacological interventions that may achieve this is physical activity. This review explores the associations between inflammation, endothelial dysfunction, and atherosclerosis and discusses the role of exercise in blocking specific pathways in the inflammation, endothelial dysfunction - atherosclerosis network.
INTRODUCTION
The endothelium is now regarded as a dynamic organ lining the entire vascular system. It is responsible for many atheroprotective functions via its regulation of the immune response, fibrinolysis, coagulation, as well as multiple other functions related to vascular growth, vasoprotection, and vasoregulation. Disruption of normal endothelial function impedes its protective effects on vascular homeostasis, an early event characterizing the development of cardiovascular disease (CVD) [1-3]. Most importantly, endothelial dysfunction and the early stages of CVD may occur in the absence of any clinical symptoms. Hence, finding methods for assessing vascular dysfunction during the early stages of the disease is important, particularly in patient groups at high CVD risk [4].
Rheumatoid arthritis (RA) is the most common chronic inflammatory arthritis characterized by symptoms and signs of joint and systemic inflammation, joint destruction, body composition alterations, and physical disability. It also associates with high CVD morbidity and mortality, which are not fully explained by the presence of traditional CVD risk factors [5-8]. In RA, increased local (synovial) and systemic expression of specific inflammatory mediators may have detrimental effects on the endothelium by enhancing biochemical processes leading to its dysfunction and eventually the development of atherosclerosis [2, 9]. Indeed, the inflammatory pathways involved in the pathogenesis of RA and atherosclerosis share many characteristics [10]. Inflammation in RA is often evaluated by measuring the acute phase reactant C-reactive protein (CRP) and/or the erythrocyte sedimentation rate (ESR), which, in combination with other clinical markers (e.g. disease activity or X-ray scores), are used to assess the disease state and response to treatment [11]. The acute phase response is attributed to increased levels of pro-inflammatory cytokines, such as tumour necrosis factor alpha (TNF-alpha) and interleukin-6 (IL-6) [10], which are individually or synergistically involved in many other disease processes, including joint destruction [12] altered body composition [13], and changes in vascular homeostasis [14, 15].
INFLAMMATION AND ENDOTHELIAL DYSFUNCTION
The endothelium controls vascular function by releasing vasoactive factors such as nitric oxide (NO), prostacyclin (PGI2), and endothelin-1 (ET-1). Endothelial dysfunction occurs when the balance between these vasoactive factors is disturbed. The down-regulation of endothelial NO synthase (eNOS) plays an important role in this process [16]. Altered endothelial homeostasis due to the disturbed NO production is characterized by diminished endothelium-dependent dilation and increased expression of adhesion molecules, as well as changes in endothelial cell phenotype and increased endothelial permeability [17]. Such changes in the structure and function of the endothelium are thought to be important as initial stages in the pathogenesis of atherosclerosis and CVD [18].
Several studies have established a strong link between inflammation and endothelial dysfunction. In vitro, CRP modulates eNOS activity of human endothelial cells [19, 20] via uncoupling eNOS and subsequently increasing superoxide anions (oxidative stress) from NADPH oxidase [21]. As a result, phosphorylation of eNOS is decreased, and this subsequently reduces bioavailability of NO [22]. Apart from NO, CRP upregulates the expression of ET-1 which is a vasoconstrictor [23]. Studies also reveal a direct effect of TNF-alpha on dowregulating the expression of eNOS and diminishing endothelial NO [15]. TNF-alpha downregulates at the same time enzymatic activities of eNOS and argininosuccinate synthetase (regulator of citrulline/NO cycle) [24] and induces an increase in mRNA degradation of both eNOS and neuronal NO synthase promoting vasoconstriction [25]. In addition, by activating NADPH oxidase, TNF-alpha stimulates the generation of reactive oxygen species within the endothelial environment further impairing NO-mediated vasodilation [26]. Apart from inhibition of NO production, TNF-alpha may also be responsible for the reduction in the bioavailability of NO [27]. This is perhaps the main reason why TNF-alpha inhibition is directly linked to improved endothelial function in both RA [28] and non-RA populations [29].
Endothelial function and morphology are reported to be significantly worse in RA compared to healthy controls and this is attributed in large part to the high-grade inflammatory state of RA [30, 31]; in fact, acute inflammation (e.g. in response to immunization) and low-grade chronic systemic inflammation (detected using high sensitivity CRP assays) in healthy individuals has been directly linked with arterial stiffness and vascular dysfunction [32, 33]. However, despite this biologically plausible link between inflammation and vascular dysfunction, there are still some controversies and inconsistent results in RA [34, 35], where some studies report a lack of association between systemic inflammatory load and endothelial function [36-38]. This may be attributed to different methodological approaches (e.g. using small, diverse patient populations cross-sectionally, or even smaller populations longitudinally but with short time courses), or lack of sufficient statistical power and no correction for multiple potential confounders (e.g. age and many CVD risk factors). The link between inflammation and vascular function appears to be more consistently demonstrated in homogeneous RA samples, such as young patients, patients with new onset RA and without established CVD [31, 39]. Vascular function may progressively deteriorate during the course of RA [40]. Hence, RA patients with long disease duration may already have significantly impaired endothelial function at the time of assessment, irrespective of disease “current” activity. Given the fluctuations in inflammation characteristic of RA, it might be that longstanding, though intermittent, inflammatory insult to the endothelium is of more importance to vascular function in RA than the current level of inflammation [41]. Studies that took into account long-term inflammatory burden have been able to demonstrate associations between this and vascular function [36, 39].
INFLAMMATION AND ATHEROSCLEROSIS
Endothelial cell dysfunction, evident as NO deficiency, appears to be amongst the earliest processes involved in atherosclerosis. Reduction of NO bioavailability disrupts the balance of the vasoactive factors, which allow ET-1 levels to increase and cause vasoconstriction [16]. Along with the biochemical mediators of endothelial function, mechanical forces and in particular, shear stress are also responsible for altering the phenotype of endothelial cells. Shear stress is the tangential stress that is applied to the artery wall and induces distension of the arteries; it is long-known that atherosclerotic lesions originate mainly in areas of low shear stress, where flow is disturbed [42, 43]. In these areas, endothelial cells promote a pro-atherogenic phenotype [44], enhancing both the local selectivity of plaque formation, as well as vessel wall remodeling, which, in turn, affects plaque vulnerability [45].
At the initial stages of atherosclerosis, leukocytes, particularly monocytes, progressively transmigrate through the endothelium with the help of monocyte chemoattractant protein 1 (MCP-1) and IL-8 [14]. Atherosclerosis progresses via the differentiation of monocytes into macrophages that retain low-density lipoproteins and become foam cells. Increased permeability of RA endothelial cells further enhances the entry of low-density lipoproteins and further promotes this cascade of events [46]. It has now been well-established that inflammation is involved in all phases of the atherothrombotic processes [2]. Whereas MCP-1 and IL-8 regulate monocyte transmigration, IL-8 can be upregulated in endothelial cells by CRP via the NF-kB pathway [47]. CRP can also increase the expression of macrophage colony stimulating factor (M-CSF), responsible for the differentiation of monocytes into macrophages. Subsequently, macrophage proliferation increases, thereby enhancing the progression of atherosclerosis via the promotion of the formation of foam cells [48]. An alternative effect of CRP is to destabilize the atheromatous plaque at later stages of atherosclerosis, by upregulation of matrix metalloproteinases [49] which causes weakening of the plaque’s, fibrous cap. Apart from the above effects of CRP on these individual steps of atherosclerosis, it also enhances fibrinolytic procedures within the endothelial environment. CRP increases plasminogen activator inhibitor-1 mRNA through induction of the NF-kB signalling pathway [50] and downregulates endothelial tissue plasminogen activator, a serine protease that is responsible for fibrinolysis. This biochemical process is orchestrated by TNF-alpha and IL-1b [51]. Other important atherogenic properties of CRP include CRP-induced oxidative stress via overproduction of reactive oxygen species [52]. Reactive oxygen species in the endothelium enhance degradation of eNOS via eNOS uncoupling and subsequent increase in superoxide [53]. In general, superoxide reacts rapidly with NO, forming peroxynitrite which significantly impairs NO bioactivity [54] and deteriorates endothelial function.
TNF-alpha has direct effects on endothelial permeability by altering the distribution of endothelial cadherin-catenin and inhibiting the re-structure of F-actin fibers [55]. This procedure may enable monocyte transmigration into the endothelium and amplify the cascade of procedures that favor the formation of foam cells from macrophages [54]. Furthermore, the proliferation of macrophages and increased formation of foam cells enhances secretion of TNF-alpha in the endothelium, perpetuating this vicious atherogenic cycle [3]. TNF-alpha also upregulates membrane expression of vascular cell adhesion molecule-1 [56], which plays a well established role in atherosclerosis [57] and it precedes the development of thickened intima with foam cell lesions [58]. Apart from these in vitro observations, increased expression of serum TNF-alpha has been found in patients with atherosclerotic complications in population studies [59, 60]. In RA patients, TNF-alpha enhances prothrombotic states such as dyslipidemia [61], but, most importantly, pooled evidence reveals that anti-TNF-alpha treatment may be in part responsible for reducing the risk for CVD events in RA [62]. Recent studies also reveal that TNF-alpha is involved in the more advanced processes of atherosclerosis, specific to formation of an advanced lesion via the inhibition of endothelial progenitor cells [63]. In RA patients, endothelial progenitor cell numbers are decreased compared to the healthy population [63], a phenomenon which reverses via blockade of TNF-alpha [64].
Inflammation in RA associates with dramatic increases of IL-6, IL-1 and TNF-alpha levels, which can trigger the acute-phase response resulting in high CRP, and have direct effects on the endothelium that may enhance atherosclerotic processes. As such, uncontrolled inflammation is thought to be amongst the prime factors involved in the accelerated atherosclerosis in RA [1, 2].
THE EFFECTS OF EXERCISE ON VASCULAR BIOLOGY
Increasing physical activity has been identified as one of the most important non-pharmacological interventions in both preventing and rehabilitating patients with non-communicable chronic diseases such as CVD. This is because exercise reverses endothelial dysfunction and has important anti-atherogenenic and anti-inflammatory effects (Fig. 1). Most importantly, even compared to standardized interventional strategies, exercise may improve survival at lower treatment costs [65].
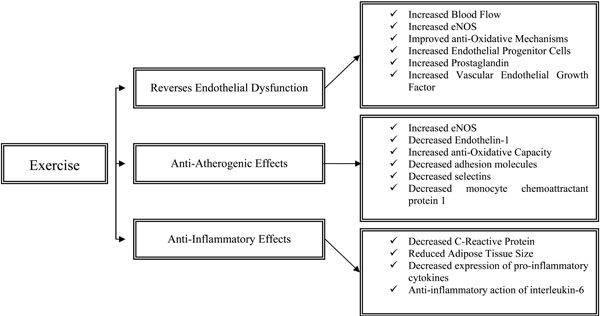
The effects of physical activity on endothelial function, atherosclerosis and inflammation.
Endothelial Function and Exercise
Exercise exerts significant effects on the endothelial system, both acutely and in the long term. These stimuli intervene and beneficially modify endothelial cell phenotype [17]. The acute effects of exercise are characterized by significant increase in blood flow pertaining to the increased metabolic demands of the exercizing muscles and are closely related to the muscle fibre type recruitment (slow or fast twitch) as well as the duration and intensity of exercise [66]. The major mechanism responsible for exercise-induced hyperaemia is endothelial NO [67] and occurs due to the increased endothelial eNOS protein expression [68] as well as eNOS activation via Akt phosphorylation [69]. Exercise acutely increases local expression of endothelial progenitor cells (endothelial repair) as well as cultured/circulating angiogenic cells which promote angiogenesis and endothelial growth [70]. Shear stress which is the result of exercise-induced blood flow enhances intracellular mechanisms which increase eNOS transcription and upregulate anti-oxidative mechanisms. Specifically, tyrosine kinase c-Src acts to increase eNOS and extracellular superoxide dismutase expression, both of which relate to better endothelial function [71]. In addition, it has been well-established that shear stress increases the expression of PG I2, a vasodilator and inhibitor of platelet aggregation [72]. Furthermore, vascular endothelial growth factor, which regulates capillary supply and exercise-induced angiogenesis, is significantly increased in response to exercise in both untrained and trained individuals [73]. In the long term, the above mentioned physiological responses promote vascular remodelling, a necessary adaptation for improved oxygen exchange and blood flow delivery [74]. These long-term improvements are characterized by improved NO bioavailability and/or endothelium-dependent vasodilation that may even reverse age-related vascular deterioration [75, 76]. Improved endothelium-dependent vasodilation in response to long-term exercise is a consistent finding in both healthy [77] and disease populations [78].
Anti-Atherogenic Effects of Exercise
Acutely, due to exercise-induced shear stress, eNOS mRNA expression increases while ET-1 decreases [79, 80], while concomitant changes involve down-regulation of vascular cell adhesion molecule 1 [81]. Oxidative stress accelerates atherogenesis via oxidation of retained low-density lipoprotein, enhancing formation of foam cells [3]. An important long-term adaptation to exercise is the improvement in the anti-oxidative capacity of the human body, i.e. resistance to oxidative stress [82]. In particular, the enzymes catalase and superoxide dismutase which decompose reactive oxygen species are significantly increased as a result of habitual physical activity [83]. Increased eNOS in the endothelium increases anti-oxidant molecules such as superoxide dismutases 1 and 3 and angiotensin receptor type 2 and decreases oxidative molecules such as NADPH oxidase and angiotensin receptor type 1 [84]. Endothelial expression of adhesion molecules, selectins and MCP-1, all of which promote atherosclerosis, also decreases [85].
Anti-Inflammatory Effects of Exercise
Physical activity and/or moderate intensity exercise have profound anti-inflammatory effects in the healthy population as well as patients with chronic diseases [86]. In population-based studies, regular physical activity is consistently associated with a reduction in CRP levels [87, 88] which is also the case in patients with RA [89, 90]. The exact mechanisms whereby regular exercise reduces CRP have not yet been fully elucidated, but it seems that this association may exist via exercise-induced reduction in hypertension, triglycerides, and apolipoproteins, factors which are directly related to CRP concentration [91]. Most importantly, exercise reduces adipose tissue which is largely responsible for secretion of IL-6, a major trigger for hepatic production of CRP [92, 93]. However, these are physiological responses of moderate physical activity/exercise, whereas strenuous exercise may acutely promote an inflammatory response. CRP may increase immediately after prolonged strenuous exercise but returns to basal levels after approximately 48 hours, while this proportionate rise in CRP depends on the intensity of the exercise and the subsequent exercise-induced muscle damage [94, 95].
Recent advances in muscle physiology reveal that muscle tissue is responsible for the production of myokines (muscle cytokines) suggesting that muscle is also an endocrine organ [96]. Muscle contractions regulate expression of specific cytokines such as IL-6, -8, -10, and -15, as well as IL-1 receptor antagonist and TNF-alpha [96-98]. It is thought that, although IL-6 is predominantly a pro-inflammatory cytokine, it may also have under certain circumstances anti-inflammatory properties [99]. In that light, and based on the fact that IL-6 increases exponentially with acute exercise up to 100 times compared to resting levels and returns to resting levels post-exercise [100], it has been suggested that IL-6 is the most important cytokine that induces the anti-inflammatory effects of exercise [96]. In contrast, pro-inflammatory cytokines IL-1b and TNF-alpha do not generally increase in response to moderate intensity exercise [101], whereas anti-inflammatory cytokines such as IL-1ra and IL-10, significantly increase [99, 102]. Interestingly, in response to regular exercise, IL-6 exerts its anti-inflammatory effects by primarily inhibiting the expression of TNF-alpha [99]. Data have demonstrated that IL-6 inhibits lipopolysacharide-induced TNF-alpha in human blood mononunclear cells [103], whereas TNF-alpha levels are overexpressed in IL-6 deficient mice [104]. Recently, the transcriptional coactivator PGC1a has also attracted attention as it suppresses various inflammatory responses and regulates the effects of exercise [105] by coactivating transcription factors involved in biogenesis of mitochondria, oxidative phosphorylation and fatty acid oxidation [106, 107]. All these biological changes in cytokine expression in response to exercise, lead to beneficial long-term effects. Indeed, a consistent finding in the literature is that regular exercise relates to decreased systemic inflammation in both healthy and diseased populations [88, 89, 108, 109].
EFFECTS OF EXERCISE ON CVD IN RA
The extensive damage of the joint structures in RA has initially led to the notion that RA patients should rest, as exercise may enhance joint damage [110]. However, during the last decade substantial evidence deriving from randomised trials reveals that exercise inhibits the progression of the disease and improves both wellbeing and functional ability of RA patients [90]. This is due to normal physiological processes which develop as a result of exercise training such as the improved muscle co-ordination and hypertrophy, reduced fat mass and better immune function. Patients that indeed have to refrain from specific exercises are those with extensive structural damage; in this occasion load of the damaged joints has to be avoided and can be replaced with alternative types of exercise. The beneficial effects of different exercise training types on disease activity and severity have been confirmed several times by various studies [110, 111] and hence, based on these evidence, exercise has now been incorporated in the management of RA [112]. It is, therefore imperative that RA patients embark on exercise training programmes. Although exercise is beneficial, increasing physical activity in this population may prove difficult [113] perhaps due to the frequent advice from rheumatology specialists to RA patients, that exercise may exacerbate disease symptoms [114]. However, data from published studies [110] of excellent quality as well as anecdotal evidence, reveal that RA patients should and can exercise at high intensities, and this is beneficial for various outcomes of this chronic disease. Moreover, studies have collectively shown that overall the adherence rates in exercise programmes can be high, presumably due to the fact that the significant improvements achieved via exercise promote self motivation [90].
In a systematic review on exercise, CVD and RA, we found that, surprisingly, no studies have investigated the effects of exercise on CVD outcomes in this population [90]. However, given the ample evidence of the beneficial effects of physical activity/exercise on inhibiting disease progression and improving disease outcomes, we have proposed a model in which exercise should be incorporated in the management of the disease in order to prevent CVD, which is highly prevalent in this population. In this model, we suggest that effective treatment and control of inflammation should precede involvement in exercise in order to prohibit further damage and discomfort for the RA patient. Various drugs are available for patients with RA, including biological agents such as TNF-alpha, which target different biological pathways for inhibiting disease progression. Overall, medication strategies are effective in ameliorating disease activity (i.e. reduced expression of pro-inflammatory cytokines and CRP), a phenomenon that is also apparent as a response to exercise training. Nevertheless, given the lack of evidence regarding the effects of exercise on RA, potential changes in medication should always be based on robust clinical outcomes and patient feedback.
Exercise involvement and/or increased physical activity are included in the management of many chronic diseases. In RA, where patients experience increased prevalence of CVD, exercise may be even more important due to its anti-atherogenic and anti-inflammatory effects [17, 99]. The majority of studies on RA and exercise investigated the effects of physical activity/exercise regimens on improving RA-related disease outcomes. We have recently shown that physically inactive RA patients have worse CVD risk profile compared to physically active patients. In this cross-sectional study, we have investigated classical and novel risk factors, the prevalence of established CVD as well as the risk assessment of developing CVDs in physically active as well as inactive RA patients. Regarding the novel CVD risk factors, our results revealed that parameters associated with vascular function (e.g. von Willebrand factor), fibrinolysis atherogenesis (lipoproteins), pro-inflammatory cytokines, and CRP were markedly improved in patients who demonstrated increased levels of physical activity [89]. However, more research is required in order to investigate the potential associations of exercise on vascular function and CVD risk in this population as well as the mechanisms that underlie these associations.
CONCLUSION
Evidence from basic research supports a clear association between inflammation, vascular dysfunction and atherosclerosis. In RA, although evidence exists to support this association for atherosclerosis, the results are equivocal at least for endothelial dysfunction, potentially due to the differences in the methodological designs that do not take into account important factors that could influence endothelial function and atherosclerosis in RA. In addition, exercise has a protective effect via inhibiting the expression of inflammatory markers and their effects on both endothelial dysfunction and promotion of atherosclerosis. Future studies in RA should specifically investigate the effects of exercise regimes on endothelial function and atherosclerosis both in population studies and at a molecular level.

